Command Palette
Search for a command to run...
Im Nature Journal Veröffentlicht! Die University of California Nutzt KI Zur Innovation Der 3D-Rekonstruktion Mittels Kryo-Elektronenmikroskopie Und Erzielt Einen Wichtigen Durchbruch in Der Strukturbiologie

Im Bereich der wissenschaftlichen Forschung geraten bestimmte Technologien aufgrund ihrer bahnbrechenden Fortschritte oft in den Fokus der Zeit. Die Kryo-Elektronenmikroskopie (Kryo-EM), die 2017 mit dem Nobelpreis für Chemie ausgezeichnet wurde, ist eine dieser Technologien. Mithilfe der Kryo-Elektronenmikroskopie gelang es Shi Yigongs Team beispielsweise im Jahr 2015 erstmals, die hochauflösende Struktur des Spleißosoms zu erfassen. Dies wurde als Chinas größter Beitrag zur Weltwissenschaft im Bereich der Biowissenschaften in den letzten 30 Jahren gefeiert und weckte auch große Aufmerksamkeit für die Kryo-Elektronenmikroskopie.
Als wichtiges Werkzeug im Bereich der Strukturbiologie kann die Kryo-Elektronenmikroskopie Proben schnell auf niedrige Temperaturen abkühlen und so die Kristallisation von Wassermolekülen in den Proben verhindern. Dadurch bleibt der nahezu physiologische Zustand der Proben erhalten.Sobald die Proben eingefroren sind, können Forscher eine Reihe von Kryo-EM-Techniken verwenden, um die Proben in 3D in verschiedenen Auflösungen, einschließlich nahezu atomarer Auflösung, zu visualisieren und so ein tieferes und umfassenderes Verständnis der Proben zu erlangen.
Obwohl die Technologie der Kryo-Elektronenmikroskopie immer ausgereifter geworden ist, war die Frage des Orientierungsvorteils bei der Probenvorbereitung immer ein schwieriges Problem. Im Allgemeinen erfordert der 3D-Rekonstruktionsprozess Proteinprojektionen aus allen Richtungen, um den gesamten Raum abzudecken. Allerdings weisen an der Luft-Flüssigkeits-Grenzfläche (AWI) adsorbierte Proteine häufig Orientierungsvorteile auf, was zu unvollständigen Projektionsdatensätzen führt, die wiederum zu unterschiedlich starken Verzerrungen der Proteindichte und damit zu Rekonstruktionsverzerrungen führen.
kürzlich,Das Forschungsteam der UCLA schlug eine selbstüberwachte Deep-Learning-Methode namens Single-Particle IsoNet (spIsoNet) vor.Diese Methode bietet eine neue Möglichkeit, die Isotropie von Proben wiederherzustellen. Wenn spIsoNet auf Einzelpartikel-Kryo-EM angewendet wird, kann es die Qualität der Rekonstruktion biologischer Makromoleküle erheblich verbessern, die Ausrichtungsgenauigkeit und Winkelisotropie erhöhen und zu neuen Durchbrüchen auf dem Gebiet der Strukturbiologie führen.
Die Forschungsarbeit mit dem Titel „Überwindung des Problems der bevorzugten Orientierung in der Kryo-EM mit selbstüberwachtem Deep Learning“ wurde in der internationalen Fachzeitschrift Nature Methods veröffentlicht.
Forschungshighlights:
* Diese Studie entwickelte eine End-to-End-Methode basierend auf selbstüberwachtem Deep Learning, spIsoNet, die zur Verbesserung der Bildqualität von Kryo-EM verwendet werden kann
* spIsoNet kann das 3D-Rekonstruktionsproblem lösen, das durch das Problem der bevorzugten Orientierung verursacht wird
* spIsoNet verbessert die Winkelisotropie und die Genauigkeit der Partikelausrichtung während der 3D-Rekonstruktion

Papieradresse:
https://doi.org/10.1038/s41592-024-02505-1
spIsoNet-Datensatzadresse:
https://go.hyper.ai/P7XQu
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Wählen Sie mehrere Datensätze mit jeweils unterschiedlichen Eigenschaften und Anwendungsszenarien aus
In dieser Studie verwendeten die Forscher mehrere Datensätze, um die Leistung von spIsoNet zu testen, jeder mit seinen eigenen einzigartigen Eigenschaften und Anwendungsszenarien:
* β-Galactosidase-Datensatz:Es enthält zwei Teilmengen mit bestimmten Ausrichtungen, nämlich 1.513 Partikel mit Seitenansicht und 950 Partikel mit Draufsicht, die verwendet werden, um zu überprüfen, ob spIsoNet die durch die bevorzugte Ausrichtung beeinflusste Bildqualität (Kartenqualität) verbessern kann.
* HA-Trimer-geneigter Datensatz (EMPIAR-10097):Dies wird durch eine Gitterneigungsstrategie erreicht, die eine geneigte Betrachtungsrichtung bietet und zur Bewertung der Fähigkeit von spIsoNet verwendet werden kann, geneigte Proben zu verarbeiten.
* Nicht verzerrter HA-Trimer-Datensatz (EMPIAR-10096):Es wurde unter gitterlosen Neigungsbedingungen gesammelt und durch den Import von 130.000 Partikeln und die Durchführung einer Fehlausrichtungskorrektur hatte das resultierende Bild eine Auflösung von 3,45 Å, die zum Vergleich der Unterschiede in den Verarbeitungseffekten zwischen geneigten und nicht geneigten Proben verwendet werden kann.
* Asymmetrischer Ribosomen-Datensatz (EMPIAR-10406):Es enthält das 70S-Ribosom des A. baumannii-Pathogens, komplexiert mit Amikacin, und wurde verwendet, um die Leistung von spIsoNet im Umgang mit komplexen biomolekularen Strukturen zu bewerten.
* HIV-VLP-Tomographie-Datensatz (EMPIAR-10164):Es enthält unreife HIV-1 dMACANC virusähnliche Partikel (VLPs) mit einer Auflösung von 3,6 Å. In dieser Studie wurde ein detaillierter Einblick in die Struktur des Viruspartikels gegeben.
spIsoNet: Basierend auf dem U-Net Deep Learning-Modell besteht es aus zwei Hauptmodulen
Das von spIsoNet verwendete neuronale Netzwerk basiert auf der U-Net-Netzwerkarchitektur.Dies ist ein Deep-Learning-Modell, das in der Wiederherstellung und Segmentierung biomedizinischer Bilder breite Anerkennung gefunden hat. Wie in Abbildung b unten dargestellt, basiert U-net auf der Encoder-Decoder-Architektur durch Stapeln von Faltungsblöcken.
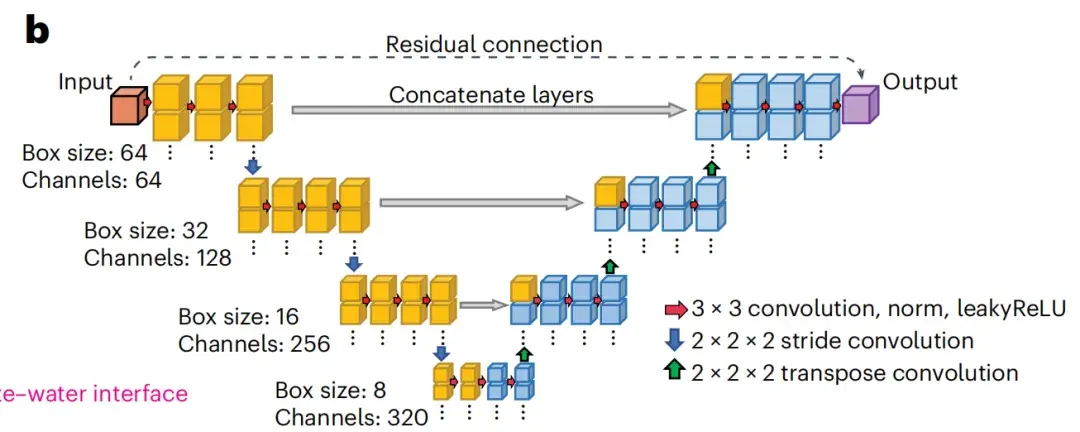
Basierend auf dem U-Net-Modell besteht spIsoNet hauptsächlich aus zwei Modulen:
Das Modul Anisotropiekorrektur
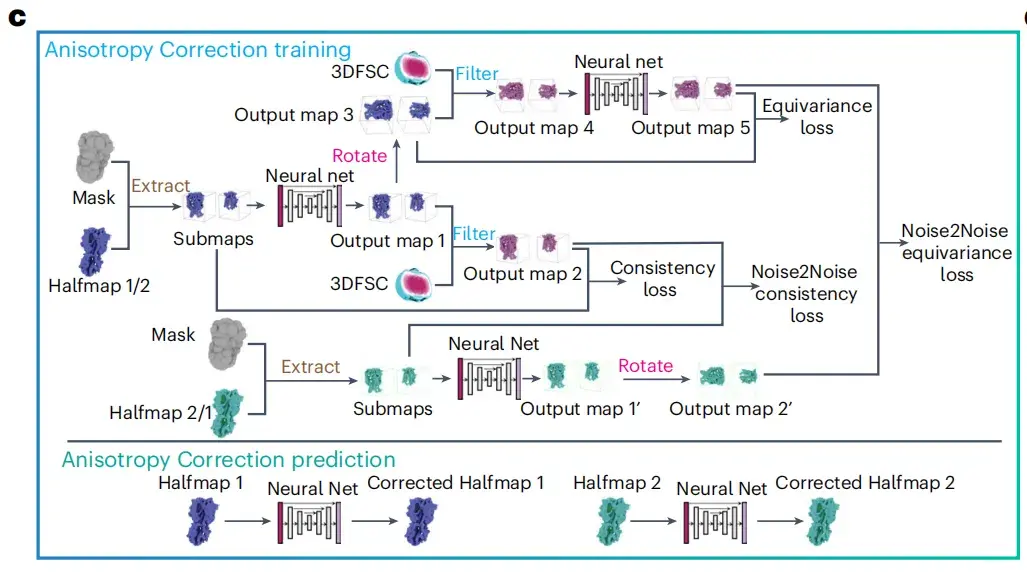
Die Forscher entwickelten ein Anisotropiekorrekturmodul, um die Klarheit von Kryo-Elektronenmikroskopiebildern zu verbessern.Wie in Abbildung c unten gezeigt, verwendet dieses Modul zwei Halbbilder, ein dreidimensionales Fourier-Shell-Korrelationsvolumen (3DFSC) und eine Lösungsmittelmaske als Eingabedaten und integriert den 3DFSC-Algorithmus, um die gewichtete Summe von vier verschiedenen Arten von Verlustfunktionen zu minimieren, darunter Konsistenzverlust, Äquivarianzverlust, Rausch-zu-Rausch-Konsistenzverlust und Rausch-zu-Rausch-Äquivarianzverlust, wodurch die Qualität der Kryo-EM-Bilder verbessert wird.
Das Modul zur Fehlausrichtungskorrektur mit Anisotropiekorrektur
Wie in Abbildung e unten dargestellt, integriert dieses Modul einen Arbeitsablauf mit drei Hauptschritten: Kartenfilterung, Anisotropiekorrektur und RELION-Autoverfeinerung. Die Anisotropiekorrektur ist der Kern des gesamten Prozesses, dessen Ziel darin besteht, die Qualität von Kryo-EM-Bildern durch Anisotropiekorrektur zu verbessern.
* Unter Anisotropiekorrektur versteht man die Korrektur der Unterschiede in den physikalischen und chemischen Eigenschaften eines Objekts in verschiedene Richtungen durch spezielle Algorithmen, um einen isotropen Effekt zu erzielen.
* Die Technologie zur Korrektur von Fehlausrichtungen wird hauptsächlich verwendet, um Bildfehlausrichtungsprobleme zu korrigieren, die durch geometrische Verzerrungen während des Bildgebungsprozesses verursacht werden.
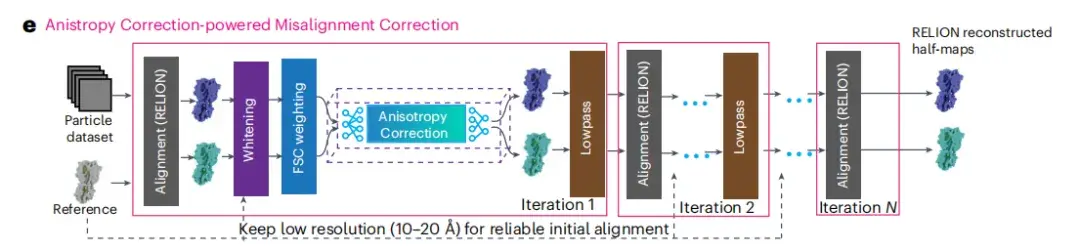
Nach Abschluss der Anisotropiekorrektur erhielten die Forscher genauere Partikelorientierungsparameter und zwei von RELION rekonstruierte Halbbilder. Nach jeder 3D-Verfeinerungsiteration werden diese Halbbilder durch Nachbearbeitungsfilter, einschließlich Weißung und FSC-Gewichtung, verarbeitet, um die Bildqualität weiter zu verbessern. Das anisotrope Korrekturmodul von spIsoNet verarbeitet dann diese gefilterten Halbbilder und die verarbeiteten korrigierten Halbbilder werden tiefpassgefiltert, um einen Standard zu erreichen, der ihrer Auflösung entspricht. Diese beiden gefilterten und korrigierten Halbbilder werden als Referenz für die anschließende Orientierungsschätzung verwendet.
Forschungsergebnisse: spIsoNet verbessert die Bildqualität von Kryo-EM deutlich
Die Anisotropiekorrektur ist effektiv
Die Forscher fanden heraus, dass das Anisotropiekorrekturmodul von spIsoNet die fehlenden Informationen in den simulierten Daten effektiv wiederherstellen kann. daher,Die Studie testete spIsoNet zunächst am RELION-Tutorial-Datensatz, der β-Galactosidase enthält.
Wie in Abbildung jm unten gezeigt, haben die Forscher durch Auswahl von Partikeln in Seitenansicht und Draufsicht aus den 2D-Klassendurchschnitten zwei Teilmengen von Partikeln mit bevorzugten Ausrichtungen zusammengestellt und eine standardmäßige RELION-3D-Rekonstruktion durchgeführt. Die Testergebnisse zeigen, dass das Anisotropiekorrekturmodul allein die 3D-Rekonstruktionsartefakte, die durch eine von oben oder von der Seite dominierte Ausrichtung verursacht werden, wirksam reduzieren kann.
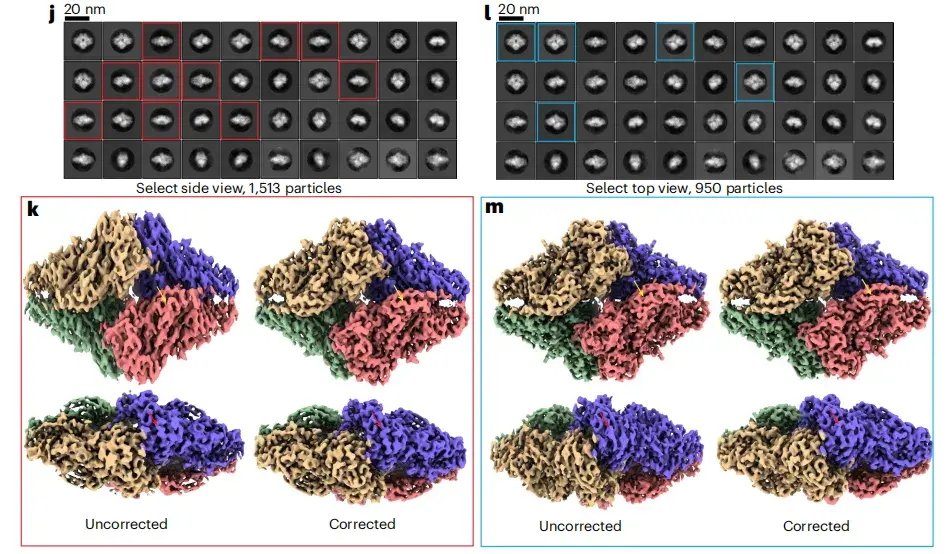
* Wobei sich jl auf die aus verschiedenen Winkeln rekonstruierte 2D-Klassifizierungskarte und km auf die aus verschiedenen Winkeln rekonstruierte 3D-Klassifizierungskarte bezieht.
Anisotropiekorrektur und Fehlausrichtungskorrektur verbessern die Qualität von Kryo-EM-Bildern deutlich
Frühere Studien haben gezeigt, dass die Kryo-EM-Bildqualität von HA-Trimer-Tilt-Datensätzen nicht ideal ist. Um die Wirkung von spIsoNet zu testen,In dieser Studie wurde erstmals eine anisotrope Korrektur an der Halbkarte durchgeführt.Die Ergebnisse zeigen, dass die Qualität des korrigierten Bildes deutlich verbessert, die lokale Auflösung erhöht und das Rauschen reduziert wird. Wie in den Abbildungen ab unten gezeigt, wird im korrigierten Bild die Seitenkettendichte deutlich sichtbar, die im Originalbild zuvor schwer zu erkennen war.
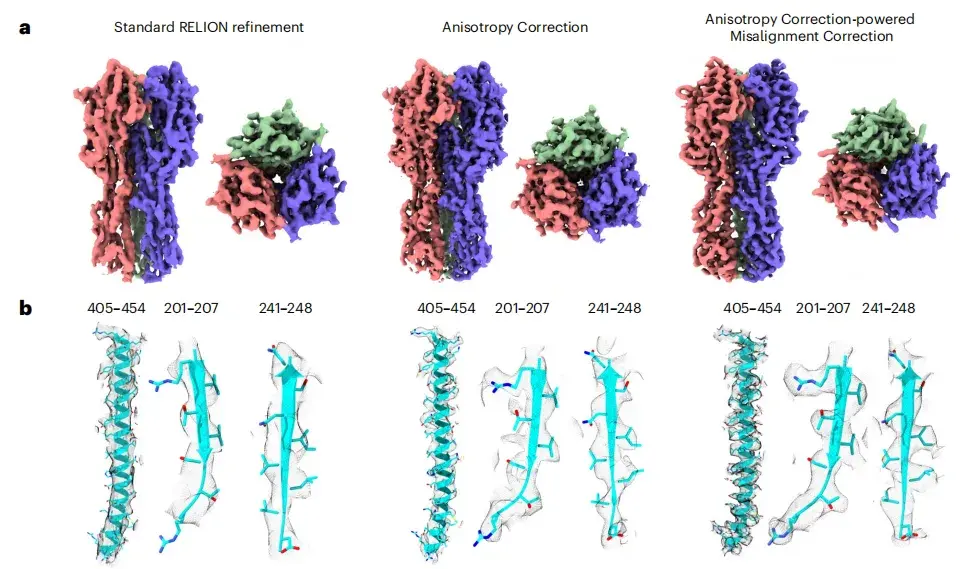
* Von links nach rechts: Standard-RELION-Verfeinerung, Anisotropiekorrektur und durch Anisotropiekorrektur gesteuerte Fehlausrichtungskorrektur.
Darüber hinaus verbessert das Bild nach der Ablenkungskorrektur, wie in Abbildung cf unten gezeigt, die Fourierschalenkorrelation vom Bild zum Modell, und die dreidimensionale Fourierschalenkorrelation (3DFSC) liegt nahe an der Kugelform (0,991). Im Vergleich zum Originalbild zeigt das Bild nach der Korrektur auch eine größere isotrope Fourier-Shell-Occupy-Area (FSO).
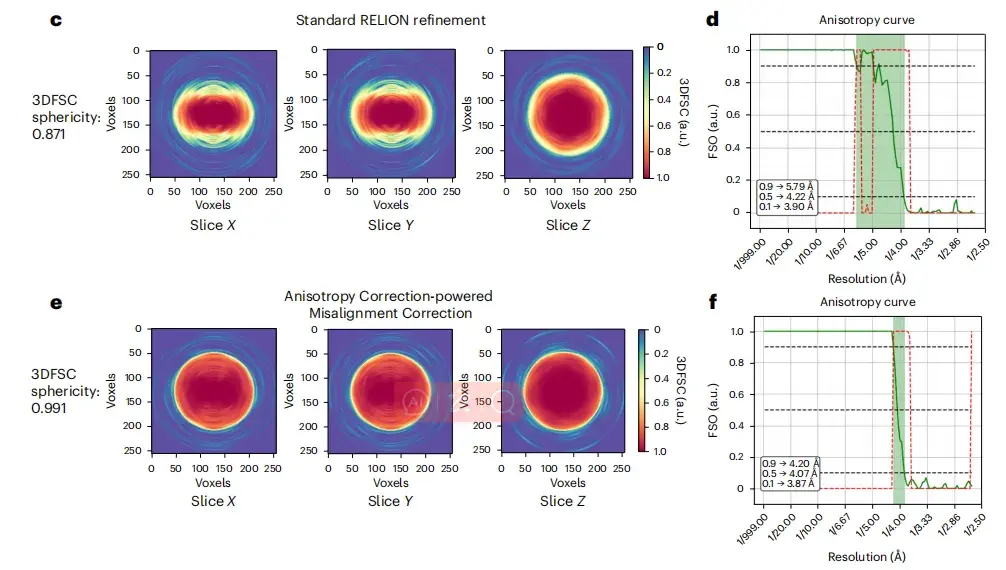
* Wobei ce die 3DFSC-Schnitte sind, die für die RELION-Verfeinerung bzw. die spIsoNet-Fehlausrichtungskorrektur verwendet werden, und df die FSO- und P-Werte des Bingham-Tests sind, die aus den Ergebnissen der RELION-Verfeinerung bzw. der spIsoNet-Fehlausrichtungskorrektur berechnet werden.
Durch die Korrektur von Fehlausrichtungen wurden viele falsch zugewiesene Richtungen erfolgreich identifiziert und korrigiert
Für einen Proteindatensatz mit schwerwiegenden Problemen bei der Orientierungsverzerrung: der nicht geneigte HA-Trimerdatensatz (EMPIAR-10096)In dieser Studie wurde das auf Anisotropiekorrektur basierende Fehlausrichtungskorrekturmodul von spIsoNet zur Verarbeitung von Partikeldatensätzen verwendet.Das rekonstruierte HA-Trimerbild im geneigten Datensatz wurde als Referenzmodell verwendet.
Nach der Korrektur der Fehlausrichtung, wie in den Abbildungen b–f unten gezeigt, erhielten die Forscher ein Bild mit der richtigen Form und erreichten eine deutliche Verbesserung der Isotropie. Wie in Abbildung h unten gezeigt, ist die durch die Halbkarten-FSC (3,5 Å) und die Modell-zu-Karte-FSC (3,6 Å) bestimmte Bildauflösung konsistent.
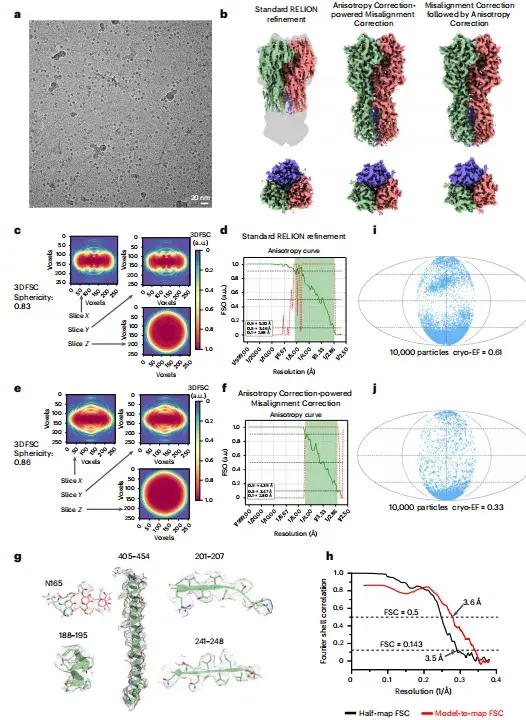
*a – repräsentative Kryo-EM-Mikrofotografien, b – Kryo-EM-Bilder des HA-Trimers, rekonstruiert mit verschiedenen Methoden, c – Schnitte von 3DFSC, verwendet für die Standard-RELION-Verfeinerung, d – FSO und P-Werte des Bingham-Tests, berechnet basierend auf den Ergebnissen der Standard-RELION-Verfeinerung, e – Schnitte von 3DFSC, korrigiert durch spIsoNet-Fehlausrichtung, f – FSO und P-Werte des Bingham-Tests, berechnet basierend auf den Ergebnissen der Korrektur der spIsoNet-Fehlausrichtung, g – repräsentative Dichte der Aminosäurereste und Glykane, ausgewählt aus Kryo-EM-Bildern, h – korrigierte FSC-Kurve des HA-Trimers, i,j – Verteilungsergebnisse in verschiedene Richtungen und entsprechende KryoEF-Werte
spIsoNet verbessert die Ausrichtung asymmetrischer Partikel und Partikel, die Nukleinsäuremoleküle enthalten
Wie in den Abbildungen ad unten gezeigt, ist die Bildqualität nach der Anisotropiekorrektur deutlich verbessert und weist eine kontinuierlichere Dichteverteilung, eine höhere lokale Auflösung und weniger Rauschstörungen auf.Die Studie ergab, dassWenn die lokale Subtomogramm-Durchschnittsbildung von 70S- oder 80S-Ribosomen als Referenz verwendet wird und die anfängliche Auflösung von 15 Å zur Ausrichtung beibehalten wird, können durchgängig qualitativ hochwertige Bilder ohne Modellverzerrung erhalten und die Auswirkungen der Anisotropie wirksam gemildert werden.
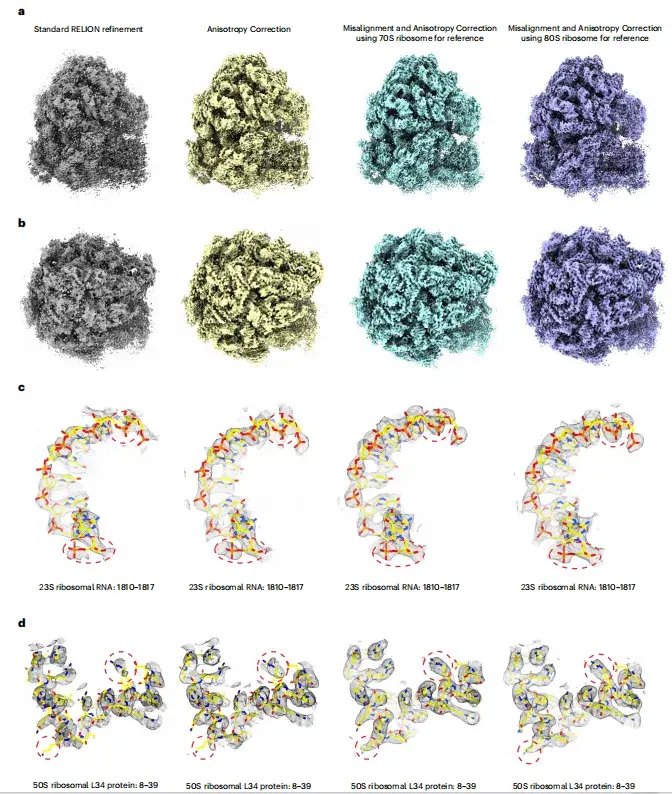
* a, b – Ribosomenbilder, rekonstruiert mit verschiedenen Rekonstruktionsmethoden, c, d – repräsentative Dichtebereiche mit angepassten Atommodellen (gelb)
spIsoNet hat Potenzial für die Anwendung in der In-situ-Strukturbiologie
Um die Anwendung von spIsoNet bei der Subtomogramm-Mittelung zu bewerten, verwendete diese Studie den HIV-1-VLP-Tomographie-Datensatz (EMPIAR-10164) als Beispiel.
Wie in Abbildung a unten gezeigt, wurde in dieser Studie im Standardprozess von RELION4 eine Teilmenge von 5 Gruppen mit unterschiedlichen Neigungswinkeln verwendet, um eine Struktur mit einer Auflösung von 3,7 Å zu erhalten. Anschließend erzielten die Forscher durch Korrektur der Fehlausrichtung eine isotrope Struktur mit einer Auflösung von 3,6 Å, wie in Abbildung e unten dargestellt.
Wie in den Abbildungen bh unten gezeigt, offenbart die Strukturanalyse außerdem eine deutlichere Seitenkettendichte und weist eine höhere 3DFSC-Sphärizität in der FSO-Kurve auf, was dazu beiträgt, die Genauigkeit der Partikelorientierungsschätzung zu verbessern.
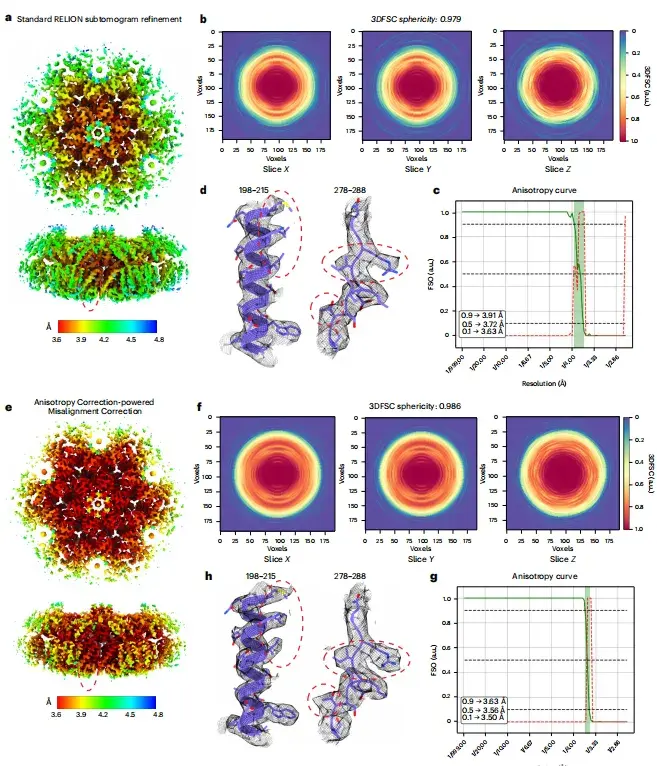
* a – Lokale Auflösungskarte von HIV-1, rekonstruiert gemäß Standard-RELION, b – Schnitt von 3DFSC, der für die Standard-RELION-Verfeinerung verwendet wurde, c – FSO und P-Wert des Bingham-Tests, berechnet gemäß den Ergebnissen der Standard-RELION-Verfeinerung, d – repräsentative Dichte von Aminosäureresten und Glykanen, ausgewählt aus Kryo-EM-Bildern, e – Lokale Auflösungskarte von HIV-1, rekonstruiert gemäß der Technologie zur Korrektur der Anisotropie von spIsoNet, f – Schnitt von 3DFSC, korrigiert durch spIsoNet-Anisotropie, g – FSO und P-Wert des Bingham-Tests, berechnet gemäß den Ergebnissen der Korrektur der Anisotropie von spIsoNet, h – repräsentative Dichte von Aminosäureresten und Glykanen, ausgewählt aus Kryo-EM-Bildern
KI+Kryo-Elektronenmikroskopie, ein technologisches Paradigma der „starken Kombination“
In den letzten zwei Jahren gab es in der wissenschaftlichen Gemeinschaft ein kontroverses Thema: „Beendet AlphaFold die Strukturbiologie?“ Die Antwort ist natürlich nein.
einerseits,Die Trainingsdaten für Strukturvorhersagemodelle wie AlphaFold stammen aus traditionellen Strukturanalysemethoden wie Röntgen- und Kryoelektronenmikroskopie.auf der anderen Seite,Die Kryo-Elektronenmikroskopie eignet sich hervorragend zur Analyse der Proteindynamik, was mit AlphaFold derzeit nicht möglich ist. Kann die KI-Technologie von AlphaFold also traditionelle Methoden der Kryo-Elektronenmikroskopie unterstützen? Man kann sagen, dass dies eine Notwendigkeit ist.
So könnten beispielsweise bereits im Jahr 2022Das Team von Professor Mao Youdong an der Peking-Universität nutzte KI + Kryo-Elektronenmikroskopie, umDie vorübergehende Umwandlung des induzierten Proteasoms von einem Zwischenzustand des Substratabbaus in einen Zwischenzustand der Substrathemmung wurde erfolgreich erfasst. Dies ist das erste Mal weltweit, dass mithilfe künstlicher Intelligenz eine Technologie zur vierdimensionalen Rekonstruktion eingesetzt wird, um die analytische Genauigkeit der zeitaufgelösten Kryo-Elektronenmikroskopie zu verbessern. Dem Team ist es gelungen, die Funktion der Dynamik von Zielproteinkomplexen, die mit schweren Erkrankungen in Zusammenhang stehen, auf atomarer Ebene zu beobachten. Die entsprechenden Ergebnisse wurden in Nature unter dem Titel „USP14-regulierte Allosterie des menschlichen Proteasoms durch zeitaufgelöste Kryo-EM“ veröffentlicht.
Link zum Artikel:
https://doi.org/10.1038/s41586-022-04671-8
Vor nicht allzu langer Zeit,Forscher des ByteDance-Forschungsteams haben eine neue Methode namens CryoSTAR vorgeschlagen.Dies ist die erste Methode, bei der modale Prioren der Proteinatomstruktur auf experimentelle Daten aus der Kryo-Elektronenmikroskopie angewendet werden. Dabei werden Informationen aus Atommodellen als Strukturregularisierung verwendet, um die Konformationsheterogenität biologischer Makromoleküle aufzuklären. Es kann grobkörnige Modelle und Dichtekarten ausgeben, um die Konformationsänderungen von Molekülen auf verschiedenen Ebenen anzuzeigen, wodurch das Anwendungspotenzial der Kryo-Elektronenmikroskopie in der dynamischen Konformationsanalyse erheblich verbessert wird. Die entsprechenden Ergebnisse wurden in Nature Methods unter dem Titel „CryoSTAR: Leveraging Structural Prior and Constraints for Cryo-EM Heterogeneous Reconstruction“ veröffentlicht.
Link zum Artikel:
https://www.nature.com/articles/s41592-024-02486-1
Es besteht kein Zweifel, dass die Kombination von KI und Kryo-Elektronenmikroskopie ein neues Kapitel in der Strukturbiologie aufschlägt und auch das große Potenzial der KI-Technologie bei der Unterstützung traditioneller Methoden der Strukturbiologie zeigt.








