Command Palette
Search for a command to run...
Teilen Sie Ihre Erfahrungen Bei Der Einreichung Bei NeurIPS 2024! Das Team Der Zhejiang-Universität Nutzt Das DePLM-Modell Zur Unterstützung Der Proteinoptimierung, Und Der Erstautor Des Artikels Demonstriert Die Demo Online

Harry Shum, ein ausländisches Mitglied der US-amerikanischen National Academy of Engineering, betonte einmal: „Wenn es etwas gibt, das wir heute tun müssen, dann ist es KI für die Wissenschaft. Es ist schwer vorstellbar, dass es heute etwas Wichtigeres gibt, und die Verleihung des Nobelpreises in diesem Jahr ist der beste Beweis dafür.“
In der Vergangenheit verließen sich Wissenschaftler auf die manuelle Datenorganisation und Hypothesen auf der Grundlage von Fachtheorien. Mithilfe der KI wird die Forschung nun direkt auf der Grundlage großer Datenmengen durchgeführt. KI für die Wissenschaft hat nicht nur die Effizienz der wissenschaftlichen Forschung verbessert, sondern auch das gesamte Paradigma der wissenschaftlichen Forschung verändert, was insbesondere im Bereich der Proteinforschung deutlich wird.
In der fünften Folge von Meet AI4S hatte HyperAI das Glück, Wang Zeyuan, einen Doktoranden vom Knowledge Engine Laboratory der Zhejiang University, einzuladen.Er gab eine ausführliche Einführung in ein Papier des für NeurIPS 2024 ausgewählten Teams mit dem Titel „Using the diffusion denoising process to help large models optimize proteins“. „DePLM: Rauschunterdrückung von Proteinsprachenmodellen zur Eigenschaftsoptimierung“.
Als führende Konferenz im Bereich KI gilt NeurIPS als eine der anspruchsvollsten, anspruchsvollsten und einflussreichsten akademischen KI-Konferenzen. In diesem Jahr gingen bei der Konferenz insgesamt 15.671 gültige Beitragseinreichungen ein, was einem Anstieg von 27% gegenüber dem Vorjahr entspricht. Die endgültige Annahmequote lag jedoch nur bei 25,8%. Die ausgewählten Arbeiten sind von großem Lernwert.In dieser gemeinsamen Sitzung stellte Dr. Wang Zeyuan das Designkonzept, die experimentellen Schlussfolgerungen, den Demo-Betriebsmodus und die Zukunftsaussichten des Rauschunterdrückungs-Proteinsprachenmodells DePLM ausführlich vor. Darüber hinaus teilte er auch seine Erfahrungen mit der Einreichung von Beiträgen bei Top-Konferenzen und hoffte, dass diese für alle hilfreich sein werden.
Dr. Wang sagte insbesondere, dass wir bei der Einreichung von Arbeiten mit der Themenauswahl, innovativen Punkten, dem Schreiben der Arbeiten und der Bewältigung interdisziplinärer Überprüfungen beginnen können.
Erstens, was die Themenauswahl betrifft,Sie können eine große Auswahl hochkarätiger Konferenzbeiträge lesen, um die wichtigeren Forschungsrichtungen zu verstehen, die die Community derzeit beschäftigen. Beispielsweise stellte Dr. Wang bei der Vorbereitung des DePLM-Papiers fest, dass Protein-Engineering, insbesondere Aufgaben zur Proteinvorhersage, bei den letztjährigen ICLR- und NeurIPS-Konferenzen ein heißes Thema war.
Zweitens, was die Innovation betrifft,Er glaubt, dass es wichtig ist, die Fähigkeit zu kultivieren, Probleme zu erkennen. Im Bereich der KI für die Wissenschaft sollten wir zunächst ein tiefgreifendes Verständnis des Wissens im Bereich der Wissenschaft erlangen und es mit den Inhalten im Bereich der KI vergleichen, um die leeren Bereiche zu ermitteln, die von der KI noch nicht erforscht wurden.
Was das Verfassen von Aufsätzen betrifft,Er sagte, der Text müsse logisch klar und detailliert sein, um sicherzustellen, dass der Artikel leicht verständlich sei. Auch ist eine verstärkte Kommunikation mit Tutoren und Kommilitonen notwendig, um nicht in eigene festgefahrene Denkmuster zu verfallen.
Schließlich wird berücksichtigt, dass AI for Science-Artikel von Gutachtern mit zwei unterschiedlichen Hintergründen geprüft werden können, wobei sich der eine mehr auf KI-Technologie und der andere mehr auf wissenschaftliche Anwendungen konzentriert.Daher ist es notwendig, beim Schreiben die Kernpositionierung des Papiers zu klären.Das heißt, unabhängig davon, ob sich dieses Papier an die KI-Community oder die Wissenschaftsgemeinschaft richtet, wird ein logischer Rahmen erstellt, um sicherzustellen, dass der Inhalt eng mit dem Thema verbunden ist.
Seiner Meinung nach hat sich der aktuelle Trend der groß angelegten Modellforschung geändert. Wir haben uns von einem einfachen Nachahmeransatz zu einem tiefen Verständnis groß angelegter Modelle entwickelt.In der Vergangenheit haben wir große Modelle an verschiedene nachgelagerte Aufgaben anpassen lassen, jetzt sind wir jedoch mehr daran interessiert, wie nachgelagerte Aufgaben besser mit der Vortrainingsphase großer Modelle zusammenarbeiten können. Je höher die Übereinstimmung zwischen beiden ist, desto besser ist die Leistung des Modells.
Beispielsweise schneiden herkömmliche, einfache Feinabstimmungsmethoden bei der Vorhersage adaptiver Landschaften hinsichtlich der Generalisierungsfähigkeit schlecht ab. Wir müssen große Modelle und unüberwachte Lernparadigmen besser verstehen, um ihre Mängel zu erkennen und sie zu verbessern. Darüber hinaus müssen wir auch auf die Mängel großer Modelle selbst achten, beispielsweise nach Möglichkeiten suchen, Modellverzerrungen zu beseitigen, um die Modellleistung zu optimieren.
Modell Open Source und testbar
Heute möchte ich ein Papier vorstellen, das wir bei NeurIPS 2024 veröffentlicht haben und in dem untersucht wird, wie das Diffusions-Rauschunterdrückungsmodell zur Unterstützung der Optimierung großer Sprachmodelle für Proteine eingesetzt werden kann.In diesem Artikel schlagen wir ein neues Denoised Protein Language Model (DePLM) vor.Der Kern dieses Ansatzes besteht darin, die vom Proteinsprachenmodell erfassten evolutionären Informationen als eine Mischung aus Informationen zu betrachten, die für die Zielmerkmale relevant und irrelevant sind, wobei irrelevante Informationen als „Rauschen“ betrachtet und eliminiert werden. Wir stellen fest, dass das vorgeschlagene rangbasierte Denoising-Verfahren die Leistung der Proteinoptimierung erheblich verbessern kann, während gleichzeitig starke Generalisierungsfähigkeiten erhalten bleiben.
Derzeit ist DePLM Open Source. Aufgrund der komplexen Konfigurationsumgebung des ModellsWir haben „DePLM: Optimieren von Proteinen mit entrauschten Sprachmodellen (kleine Beispiele)“ im Tutorial-Bereich der offiziellen HyperAI-Website veröffentlicht.Damit Sie unsere Arbeit besser verstehen und reproduzieren können, erkläre ich Ihnen die Funktionsweise des Modells aus verschiedenen Blickwinkeln. Dazu gehören die Ausführung des DePLM-Modells, die zugehörigen Konfigurationsdateien, die Feinabstimmung der Diffusionsschritte des Modells und die Ausführung des DePLM-Modells mit Ihrem eigenen Datensatz.
DePLM Open Source-Adresse:
https://github.com/HICAI-ZJU/DePLM
Adresse des DePLM-Tutorials:
Hintergrund: Maximierung der Nutzung evolutionärer Informationen und Minimierung der Einführung von Datenverzerrungssignalen
Das Forschungsobjekt dieses Artikels ist Protein, ein biologisches Makromolekül, das aus 20 Aminosäuren in einer Reihe besteht. Es erfüllt Funktionen wie Katalyse, Stoffwechsel und DNA-Replikation im Körper und ist außerdem der Hauptausführer aller Lebensaktivitäten. Biologen unterteilen seine Struktur üblicherweise in vier Ebenen. Die erste Ebene beschreibt, wie Proteine zusammengesetzt sind; die zweite Ebene beschreibt die lokale Struktur von Proteinen, wie etwa häufige α-Helices und β-Faltungen; die dritte Ebene beschreibt die gesamte dreidimensionale Struktur von Proteinen; und die vierte Ebene betrachtet die Wechselwirkungen zwischen Proteinen.
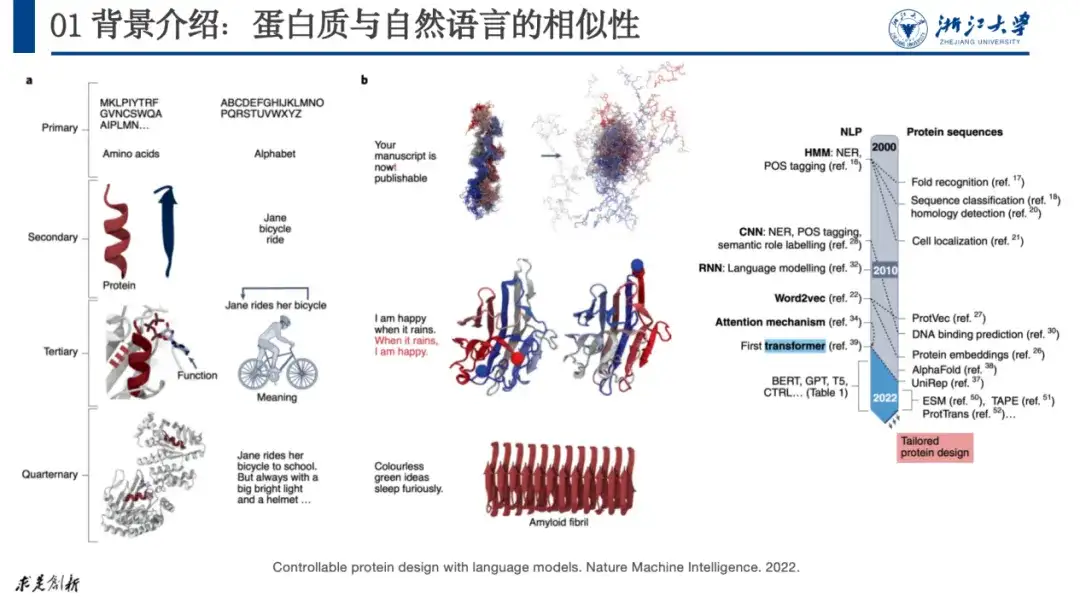
Derzeit lässt sich der Großteil der KI+Protein-Forschung auf die Forschung zur natürlichen Sprachverarbeitung zurückführen, da zwischen beiden Gemeinsamkeiten bestehen. Beispielsweise können wir die Quartärstruktur von Proteinen mit Buchstaben, Wörtern, Sätzen und Absätzen in der natürlichen Sprache vergleichen. Wenn in einem Satz ein Buchstabenfehler auftritt, verliert der Satz seine Bedeutung. Ebenso kann eine Mutation in den Aminosäuren eines Proteins dazu führen, dass das Protein keine stabile Struktur mehr bilden kann und somit seine Funktion verliert.
Wie in der folgenden Abbildung gezeigt, korrelierten Forscher in der Arbeit „Controllable protein design with language models“ natürliche Sprache mit Proteinen. Dieser Ansatz wird von Forschern weitgehend anerkannt. Seit 2020 verzeichnet die Proteinforschung ein explosives Wachstum.
Originalarbeit:
https://www.nature.com/articles/s42256-022-00499-z
Das Thema, das wir dieses Mal diskutieren, ist die KI+Protein-Optimierung, das heißt, wenn wir ein Protein haben, das nicht wie erwartet funktioniert, wie können wir seine Aminosäuresequenz anpassen, damit es die erwartete Funktion erfüllt.
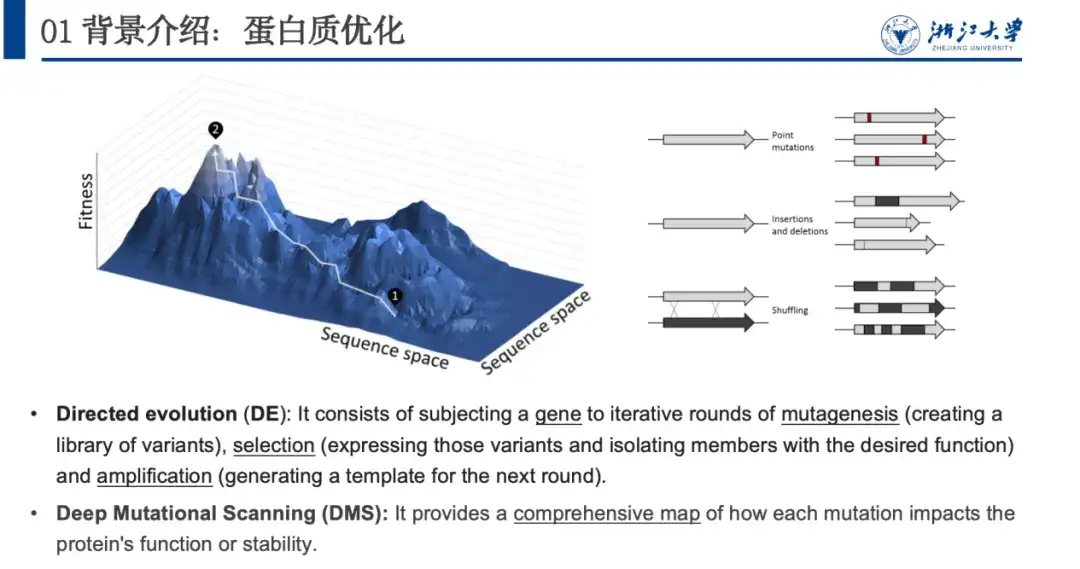
In der Natur optimieren sich Proteine kontinuierlich durch zufällige Veränderungen, darunter Punktinsertionen, -deletionen oder -mutationen. Durch Nachahmung dieses Prozesses schlugen Biologen eine gerichtete Evolution und ein tiefes Mutations-Scanning zur Optimierung von Proteinen vor. Das Problem bei diesen beiden Methoden besteht darin, dass sie zu viele experimentelle Ressourcen verbrauchen. daher,Wir verwenden rechnergestützte Methoden, um die Beziehung zwischen Proteinen und ihrer Eigenschaftsfitness zu modellieren, d. h., um die Fitnesslandschaft vorherzusagen, die für die Proteinoptimierung entscheidend ist.
Um dieses Problem zu modellieren, verwenden wir normalerweise Datensätze, Bewertungsmetriken und Berechnungsmethoden.Wie in der folgenden Abbildung gezeigt, enthält ein Proteinoptimierungsdatensatz normalerweise eine Wildtypsequenz xwt, mehrere Mutationspaare μi und den vorhergesagten Fitnesswert yi nach der Mutation. Bewertungsmodelle basieren hauptsächlich auf dem Korrelationskoeffizienten nach Spearman. Bei diesem Indikator steht nicht der konkrete vorhergesagte Wert im Vordergrund, sondern die Rangfolge der durch Mutationen verursachten Fitnesswertänderungen. Je näher der Rangwert der tatsächlichen Mutation R(Y) am vorhergesagten Fitnesswert liegt, desto besser ist das Modelltraining.
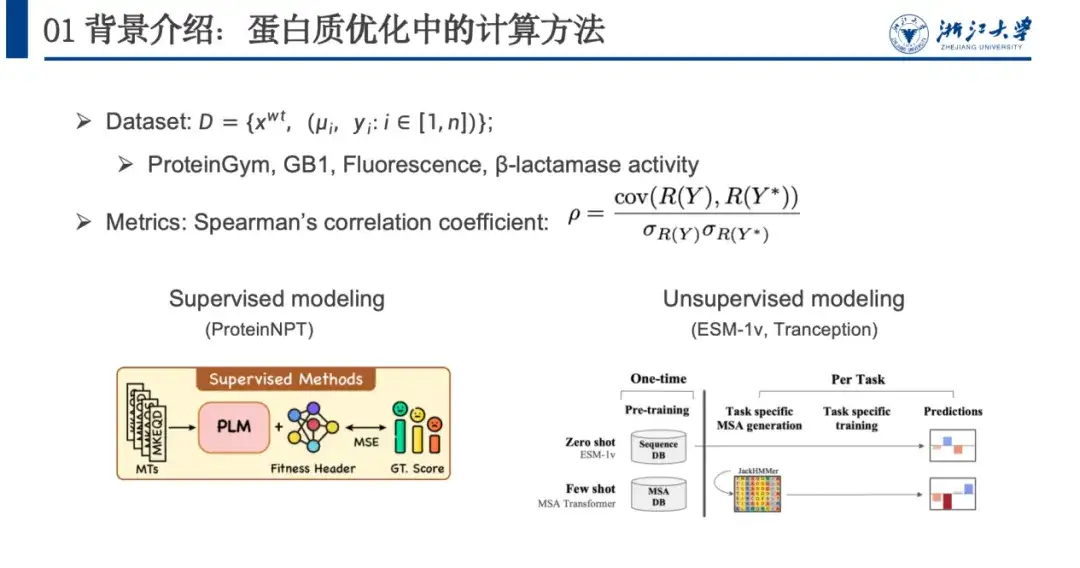
Rechenmethoden können grob in überwachte und unüberwachte Modellierung unterteilt werden. Überwachtes Lernen basiert auf gekennzeichneten Daten und trainiert das Modell durch Optimierung der Verlustfunktion, um die Vorhersagefähigkeit der Fitness zu verbessern. Für das unüberwachte Lernen sind keine gekennzeichneten Daten erforderlich, sondern es wird selbstüberwachtes Lernen anhand eines umfangreichen Proteindatensatzes durchgeführt, der nichts mit der Fitness zu tun hat. Das Modell muss nur einmal trainiert werden und kann auf verschiedene Aufgaben zur Proteinvorhersage verallgemeinert werden.
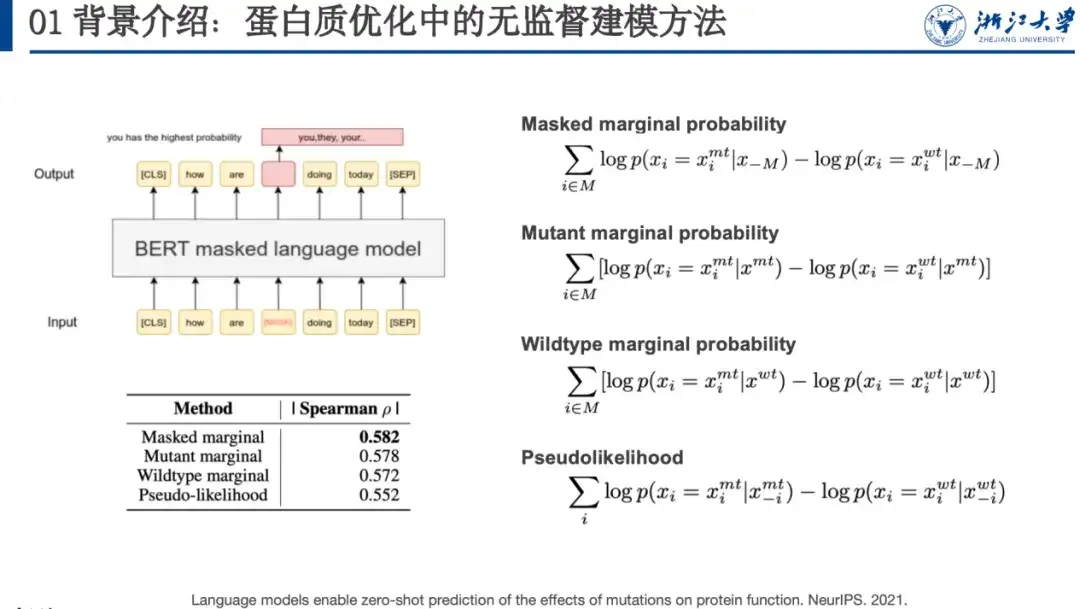
Beispielsweise ist maskierte Sprachmodellierung eine Methode des unüberwachten Lernens. Beim Trainieren des Modells müssen wir dem Modell eine kontaminierte Sequenz bereitstellen. Wir können ein Wort maskieren (wie das Wort im roten Kästchen in der Abbildung unten) oder es zufällig in ein anderes Wort ändern und es vom Sprachmodell wiederherstellen lassen, d. h. die ursprüngliche Sequenz wiederherstellen. In einem Artikel auf der NeurIPS 2021 stellten Forscher fest, dass die Wahrscheinlichkeit einer von solchen Modellen vorhergesagten Proteinmutation mit der Fitnesslandschaft korreliert. Zu diesem Zweck entwickelten sie vier Formeln zur Mutationsbewertung, wie auf der rechten Seite der folgenden Abbildung dargestellt.
Originalarbeit:
https://proceedings.neurips.cc/paper/2021/file/f51338d736f95dd42427296047067694-Paper.pdf
Zusammenfassend lässt sich sagen, dass überwachte Methoden zwar eine gute Leistung erbringen, jedoch nur begrenzte Generalisierungsfähigkeiten besitzen, während unüberwachte Methoden etwas schlechtere Leistungen erbringen, jedoch über starke Generalisierungsfähigkeiten verfügen.Um die Vorteile beider Methoden zu kombinieren, haben wir, wie in der folgenden Abbildung gezeigt, die Strategie „Vorabtraining + Feinabstimmung“ aus dem NLP-Bereich übernommen. Nach einigen Versuchen stellten wir fest, dass diese Methode zwar gute Ergebnisse lieferte, jedoch eine schlechte Generalisierungsfähigkeit aufwies, ähnlich wie beim überwachten Lernen. Anschließend analysierten wir, warum unüberwachte Methoden über hervorragende Generalisierungsfähigkeiten verfügen, und stellten die Hypothese auf, dass diese Generalisierungsfähigkeit auf evolutionären Informationen (EI) beruht. Denn Organismen können durch natürliche Evolution Proteine optimieren und solche evolutionären Mutationen bleiben auch erhalten. Daher gehen wir davon aus, dass die Korrelation zwischen Mutationswahrscheinlichkeit und Fitnesslandschaft positiv korreliert ist.

Wenn wir jedoch versuchen, das Modell zu optimieren, verwenden wir tatsächlich die Einbettungsinformationen und nutzen die Evolutionsinformationen nicht vollständig. Darüber hinaus sind die Nassversuchsdaten durch irrelevante Informationen verzerrt. Wir glauben, dass evolutionäre Informationen umfassende Informationen in verschiedene Richtungen enthalten, wie etwa Stabilität, Aktivität, Expression, Bindung usw. Wenn wir die Stabilität von Proteinen optimieren, ist die Evolution von Aktivität, Expression und Bindung eine irrelevante Information. Wenn der Wahrscheinlichkeitswert dieser uninteressanten Informationen entfernt werden kann, kann die Leistung des Modells verbessert werden. Da der gesamte Prozess im Wahrscheinlichkeitsraum ausgeführt wird, hat dies keinen Einfluss auf die Generalisierungsfähigkeit des Modells.Daher müssen wir bei der Feinabstimmung die Nutzung evolutionärer Informationen maximieren und gleichzeitig das in den Datensatz eingeführte Bias-Signal minimieren.
DePLM-Algorithmus-Framework: Rauschunterdrückungsmodell basierend auf dem Sortierraum
Darauf aufbauend haben wir das DePLM-Modell vorgeschlagen, dessen Kernidee darin besteht, die vom Proteinsprachenmodell erfassten Evolutionsinformationen als eine Fusion interessanter und uninteressanter Signale zu betrachten. Letzteres wird bei der Optimierung der Zielattribute als „Rauschen“ angesehen und muss eliminiert werden. DePLM beseitigt Rauschen in evolutionären Informationen, indem es einen Diffusionsprozess im Ordinationsbereich der Attributwerte durchführt, wodurch die Generalisierungsfähigkeit des Modells verbessert und Mutationseffekte vorhergesagt werden.
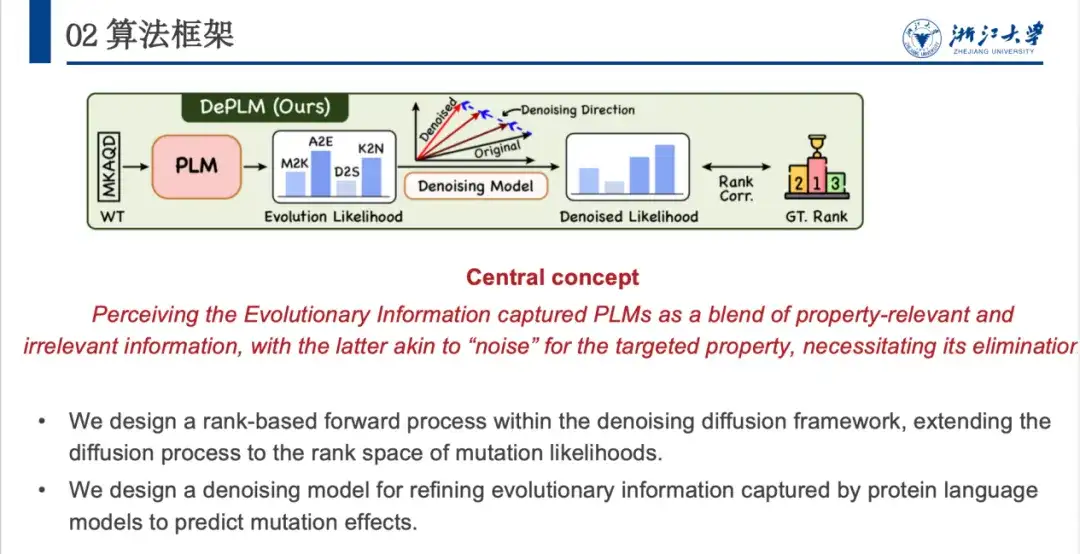
Anhand der Aminosäuresequenz eines Proteins sagt das Modell die Wahrscheinlichkeit voraus, mit der jede Position zu verschiedenen Aminosäuren mutiert. Aus der evolutionären Wahrscheinlichkeit wird dann über das Denosing-Modul die Wahrscheinlichkeit der betreffenden Eigenschaft abgeleitet. Speziell,DePLM besteht hauptsächlich aus zwei Teilen: dem Vorwärtsdiffusionsprozess und dem erlernten Rückwärtsrauschenprozess.Im Vorwärtsprozess wird der tatsächlichen Situation allmählich eine kleine Menge Rauschen hinzugefügt, und im Rückwärts-Rauschunterdrückungsprozess wird gelernt, das angesammelte Rauschen allmählich zu entfernen und die tatsächliche Situation wiederherzustellen.
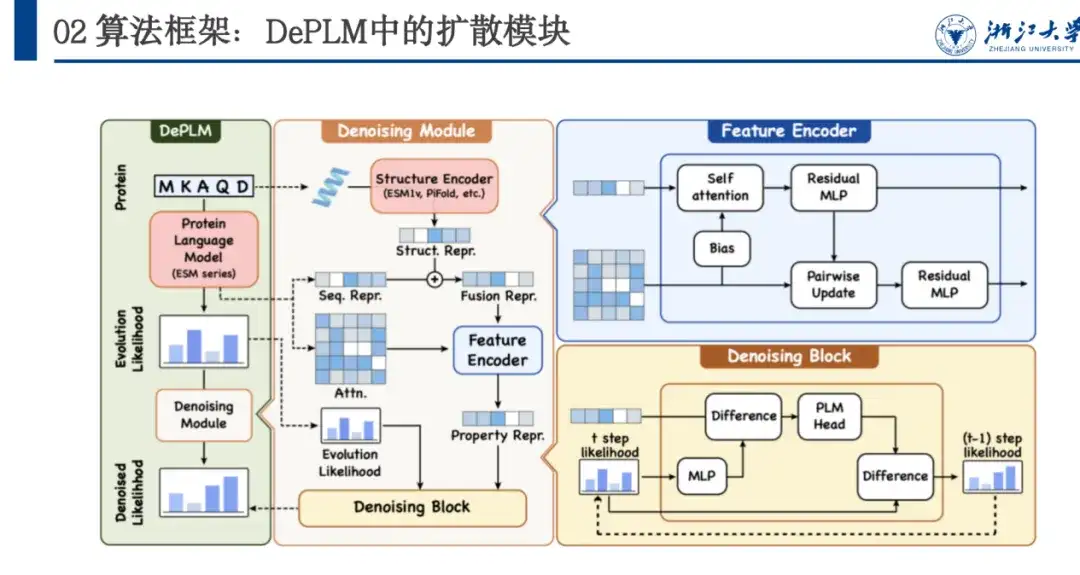
Wie in der folgenden Abbildung dargestellt, basiert DePLM auf der ESM-Serie und übernimmt die Transformer-Architektur. Sein Denosing-Modul basiert auf dem Training von Diffusionsprozessen und die Netzwerkarchitektur umfasst Feature Encoder und Denosing Block. Feature Encoder extrahiert Sequenzmerkmale aus dem Protein Language Model und extrahiert Strukturmerkmale über das ESM 1v-Modell. Diese beiden Merkmale werden als Ankerpunkte verwendet und es werden mehrere Runden von Denoising-Block-Iterationen verwendet, um schrittweise das Rauschen zu beseitigen und die Denosed-Wahrscheinlichkeit zu erhalten.
In der Vergangenheit wurden Denoising-Methoden vor allem im Bereich der Bilderzeugung, insbesondere bei Diffusionsmodellen, eingesetzt. Wie in der folgenden Abbildung gezeigt, wird das Originalbild x0 durch einen definierten Rauschunterdrückungsprozess in einen Rauschraum (xT) nahe einer Gauß-Verteilung umgewandelt, und dann lernt das Modell den umgekehrten Rauschunterdrückungsprozess.
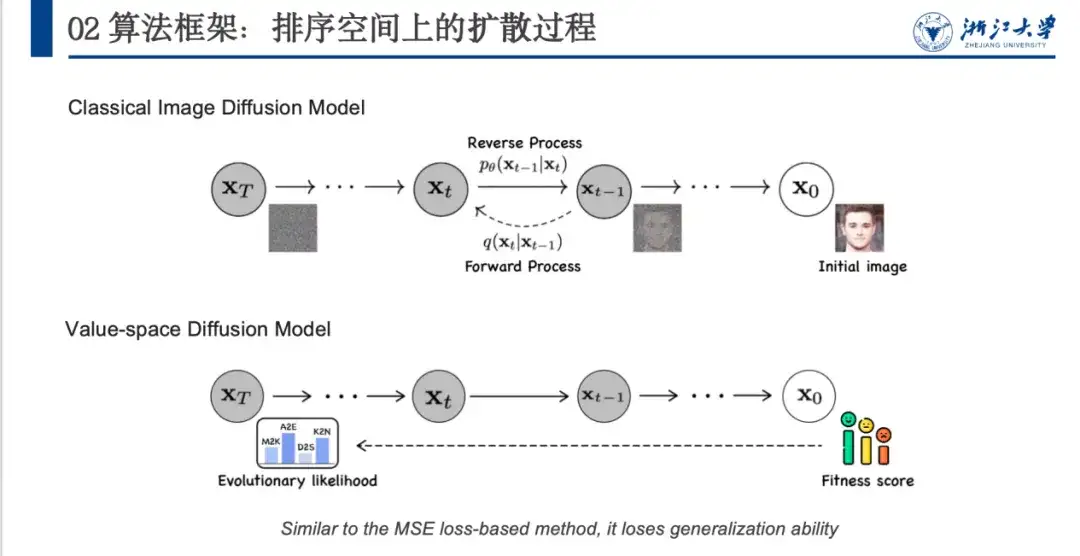
Allerdings gibt es einige Probleme bei der direkten Anwendung von Bildrauschminderungsmodellen auf den Proteinbereich. Wie in der obigen Abbildung gezeigt, kann das Bildrauschenmodell zufälliges Rauschen hinzufügen, um einen untrennbaren Rauschraum (von x0 bis xT) zu bilden. Proteine verfügen jedoch über einen Fitnesswert und eine evolutionäre Wahrscheinlichkeit und der Anfangs- und Endzustand sind festgelegt. Daher muss der Rauschadditionsprozess sorgfältig geplant werden. Zweitens wird sich das Modell am Fitness-Score ausrichten, was zwar zu einer guten Leistung, aber einer schlechten Generalisierungsfähigkeit führt.
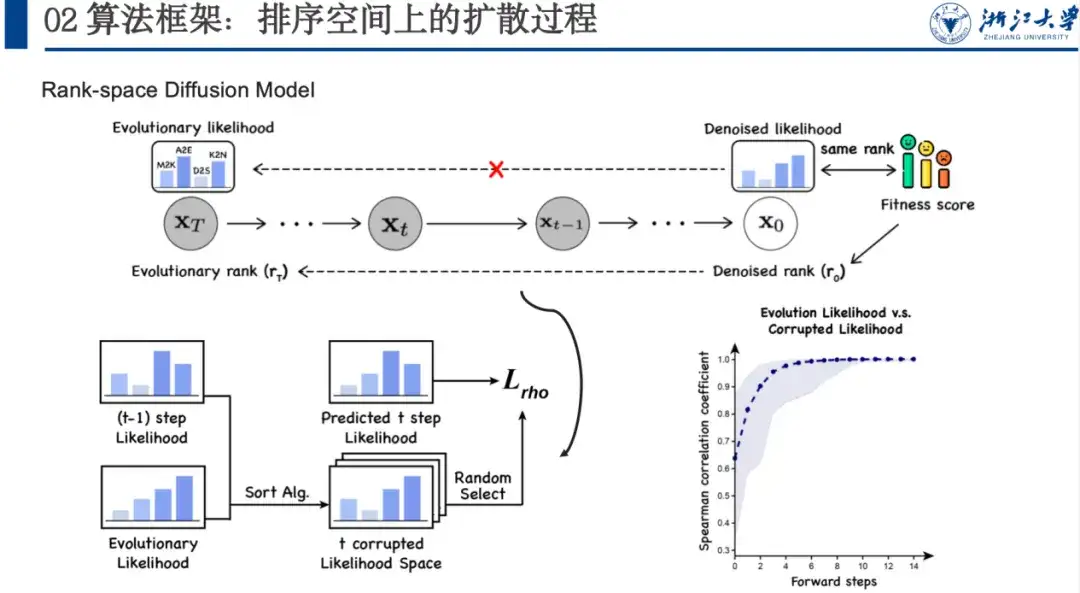
Wir schlagen daher ein Rauschunterdrückungsmodell auf Grundlage des Ranking-Raums vor, dessen Schwerpunkt auf der Maximierung der Ranking-Relevanz liegt.Der Grund hierfür ist, dass wir die evolutionäre Wahrscheinlichkeit für den Eigenschaftsraum, der von Interesse ist, entrauschen möchten. Obwohl wir die spezifische Situation dieses Raums nicht kennen, wissen wir, dass seine Sortierung mit der Fitnesssortierung übereinstimmt.
Wir fügen diesem Raum Rauschen hinzu und lassen das Modell eine große Anzahl von Datensätzen lernen, wobei es schrittweise lernt, wie die Denosied-Wahrscheinlichkeit aussehen sollte, anstatt den Fitnesswert direkt anzupassen. In diesem vorwärts gerichteten, verrauschten Prozess verwenden wir einen Sortieralgorithmus, um jeden Sortierschritt näher an den Endzustand heranzuführen und Zufälligkeit einzudämmen. Das Modell lernt auch die Idee der umgekehrten schrittweisen Sortierung. Insbesondere können wir, wie in der folgenden Abbildung gezeigt, wenn wir xt-1 haben, xt-1 und xT in den Sortieralgorithmus einspeisen und ihn mehrere Male sortieren lassen. Nachdem wir den Sortierraum des t-ten Schritts erhalten haben, können wir die Sortiervariablen des t-ten Schritts zufällig daraus auswählen, das Modell die Wahrscheinlichkeit vom t+1-Schritt bis zum t-ten Schritt vorhersagen lassen und den Spearman-Verlust berechnen. Da wir nicht viele Schritte wie die Bildentrauschung hinzufügen müssen, kann der Sortiervorgang normalerweise in 5–6 Schritten abgeschlossen werden, was auch die Effizienz verbessert.
Experimentelles Fazit: DePLM verfügt über eine überlegene Leistung und eine starke Generalisierungsfähigkeit
Um die Leistung von DePLM bei Protein-Engineering-Aufgaben zu bewerten, vergleichen wir es mit von Grund auf trainierten Protein-Sequenz-Encodern, selbstüberwachten Modellen usw. auf den ProteinGym-, β-Lactamase-, GB1- und Fluoreszenz-Datensätzen. Die Ergebnisse sind in der folgenden Abbildung dargestellt. DePLM übertrifft das Basismodell.Wir stellen fest, dass qualitativ hochwertige evolutionäre Informationen die Feinabstimmungsergebnisse erheblich verbessern können, was die Wirksamkeit unseres vorgeschlagenen Denoising-Trainingsverfahrens demonstriert und den Vorteil der Integration evolutionärer Informationen mit experimentellen Daten bei Protein-Engineering-Aufgaben bestätigt.
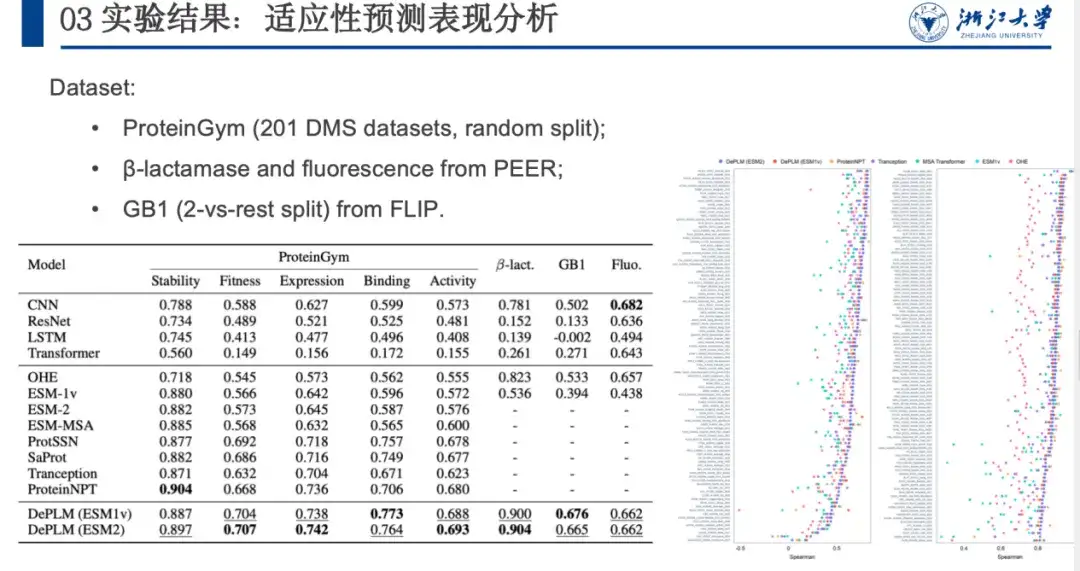
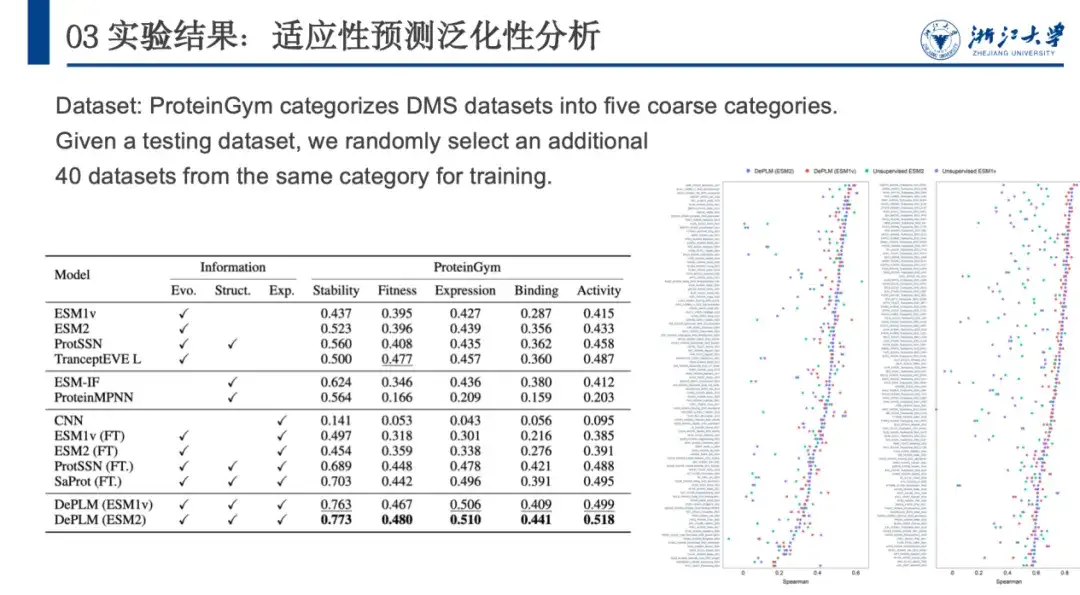
Um die Generalisierungsfähigkeit von DePLM weiter zu bewerten, klassifizierte ProteinGym den DMS-Datensatz anschließend entsprechend der gemessenen Proteineigenschaften in fünf Kategorien, nämlich Stabilität, Fitness, Expression, Bindung und Aktivität. Wir vergleichen es mit anderen selbstüberwachten Modellen, strukturbasierten Modellen und überwachten Basismodellen. Die Ergebnisse sind in der folgenden Abbildung dargestellt. DePLM übertrifft alle Basismodelle.Dies zeigt, dass Modelle, die sich nur auf ungefilterte evolutionäre Informationen stützen, unzureichend sind, da sie durch die gleichzeitige Optimierung mehrerer Ziele häufig die Zielattribute verwässern. Durch die Eliminierung des Einflusses irrelevanter Faktoren verbessert DePLM die Leistung erheblich.
Um die Generalisierungsleistung weiter zu analysieren und die Bedeutung des Herausfilterns attributirrelevanter Informationen zu bestimmen, haben wir eine Kreuzvalidierung des Trainings und der Tests zwischen den Attributen durchgeführt. Wie in der folgenden Abbildung gezeigt, ist die Leistung des Modells in den meisten Fällen geringer, wenn es auf Attribut A trainiert und auf Attribut B getestet wird, als wenn es auf dasselbe Attribut (d. h. A) trainiert und getestet wird.Dies zeigt, dass die Optimierungsrichtungen verschiedener Eigenschaften nicht konsistent sind und es zu gegenseitigen Interferenzen kommt, was unsere ursprüngliche Hypothese bestätigt.
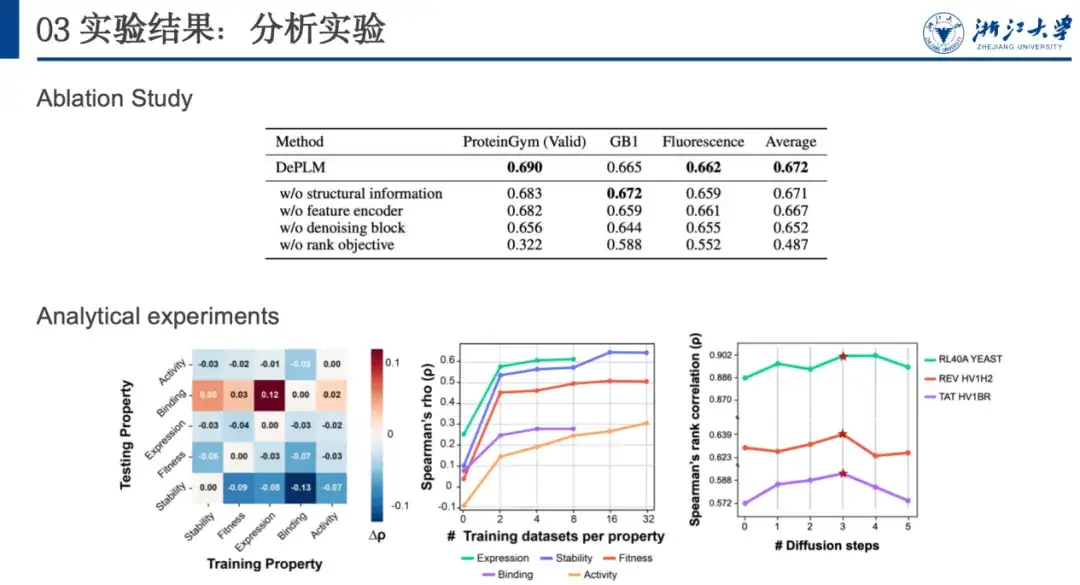
Darüber hinaus stellten wir fest, dass die Modellleistung durch das Training mit Datensätzen anderer Eigenschaften und das Testen mit dem Binding-Datensatz verbessert wurde. Dies kann auf das begrenzte Datenvolumen und die geringe Datenqualität des Binding-Datensatzes zurückgeführt werden, was zu seiner eigenen unzureichenden Generalisierungsfähigkeit führt. Das inspiriert uns,Wenn bei der Optimierung von Proteinen mit neuen Eigenschaften weniger Datensätze zu dieser Eigenschaft vorliegen, kann man die Verwendung von Daten mit verwandten Eigenschaften zur Rauschunterdrückung und zum Training in Betracht ziehen, um bessere Generalisierungsmöglichkeiten zu erhalten.
Den Bereich Protein weiter vertiefen
Gast dieser Live-Übertragung ist Wang Zeyuan, Doktorand am Knowledge Engine Laboratory der Zhejiang-Universität. Sein Team unter der Leitung von Professor Chen Huajun, dem Forscher Zhang Qiang und anderen widmet sich der akademischen Forschung in den Bereichen Wissensgraphen, große Sprachmodelle, KI für die Wissenschaft usw. und hat zahlreiche Artikel auf führenden KI-Konferenzen wie NeurIPS, ICML, ICLR, AAAI und IJCAI veröffentlicht.
Zhang Qiangs persönliche Homepage:
https://person.zju.edu.cn/H124023
Im Proteinbereich hat das Team nicht nur fortschrittliche Modelle wie DePLM zur Optimierung von Proteinen vorgeschlagen, sondern auch daran gearbeitet, die Lücke zwischen biologischen Sequenzen und der menschlichen Sprache zu schließen.Zu diesem Zweck schlugen sie das InstructProtein-Modell vor.Verwenden Sie Wissensanweisungen, um die Proteinsprache und die menschliche Sprache aufeinander abzustimmen, die bidirektionalen Generierungsfunktionen zwischen der Proteinsprache und der menschlichen Sprache zu erkunden, biologische Sequenzen in große Sprachmodelle zu integrieren und die Lücke zwischen den beiden Sprachen effektiv zu schließen. Experimente mit einer großen Anzahl bidirektionaler Protein-Text-Generierungsaufgaben zeigen, dass InstructProtein die Leistung bestehender hochmoderner LLMs übertrifft.
Klicken Sie hier, um weitere Details anzuzeigen: Ausgewählt für die ACL2024-Hauptkonferenz | InstructProtein: Angleichung der Proteinsprache an die menschliche Sprache durch Wissensanweisungen
Darüber hinaus schlug das Team auch eine Mehrzweckmethode zum Protein-Sequenz-Design namens PROPEND vor, die auf dem „Pre-Training and Prompting“-Framework basiert.Durch Eingabeaufforderungen an Skelette, Blaupausen, Funktionsbezeichnungen und deren Kombinationen können verschiedene Eigenschaften direkt gesteuert werden, und diese Methode ist äußerst praktisch und genau. Unter den fünf in In-vitro-Experimenten getesteten Sequenzen erreichte PROPENDs maximale funktionelle Wiederherstellungsrate 105,21 TP3T und übertraf damit die 50,81 TP3T der klassischen Design-Pipeline deutlich.
Originalarbeit:
https://www.biorxiv.org/content/10.1101/2024.11.17.624051v1
Derzeit sind viele der vom Team veröffentlichten Ergebnisse Open Source. Darüber hinaus stellen sie herausragende Postdoktoranden, 100 Mitarbeiter, F&E-Ingenieure und andere Vollzeitforscher auf langfristiger Basis ein. Jeder ist herzlich eingeladen mitzumachen~
Github-Homepage des Labors:
http://github.com/zjunlp









