Command Palette
Search for a command to run...
Im Science Journal Veröffentlicht! Die Shanghai Jiao Tong University Und Das Shanghai AI Lab Haben Gemeinsam Ein Proteinmutanten-Designmodell Veröffentlicht, Das Die Fortschrittlichsten Methoden Übertrifft

Proteine sind nicht nur die Antriebskräfte menschlicher Lebensaktivitäten, sondern spielen auch in vielen Bereichen eine wichtige Rolle, beispielsweise in der Biomedizin, der Lebensmittelverarbeitung, der Brauindustrie, der chemischen Industrie usw. Daher wird ununterbrochen an der Struktur und Funktion von Proteinen geforscht, um Proteine auszuwählen, die den Anforderungen für industrielle Anwendungsszenarien entsprechen und eine hohe Stabilität aufweisen.
Allerdings sind die physikalischen und chemischen Bedingungen (wie Temperatur und pH-Wert), die für die Funktionsfähigkeit von aus Organismen extrahierten „Wildtyp“-Proteinen in industriellen Umgebungen erforderlich sind, meist weit von ihrer natürlichen biologischen Umgebung entfernt. Mit anderen Worten: Aufgrund der Stabilität dieses Proteintyps ist seine Anpassung an raue Industrieumgebungen schwierig. Um den Anforderungen verschiedener Anwendungsszenarien gerecht zu werden,Mutationen sind häufig erforderlich, um die physikochemischen Eigenschaften von Proteinen zu verbessern und dadurch ihre Stabilität unter extremen Temperatur-/pH-Bedingungen zu erhöhen oder die Enzymaktivität und -spezifität zu steigern.
Es ist zu beachten, dass die Veränderung der biologischen Aktivität eines Proteins jahrelange experimentelle Forschung zu seinem Wirkungsmechanismus erfordert, was nicht nur zeitaufwendig und mühsam ist, sondern es wird auch immer schwieriger, den sich schnell ändernden Modifikationsanforderungen gerecht zu werden. In den letzten Jahren hat sich durch die Einführung von Proteinsprachenmodellen die Genauigkeit der Proteinfitnessvorhersage erheblich verbessert, die Genauigkeit der Stabilitätsvorhersage ist jedoch noch immer unzureichend.
Wirklich bedeutsame Proteinmutationen sollten die Stabilität verbessern und gleichzeitig ihre biologische Aktivität erhalten und umgekehrt. Als Reaktion darauf haben die Forschungsgruppe von Professor Hong Liang an der School of Natural Sciences/School of Physics and Astronomy der Shanghai Jiao Tong University zusammen mit Tan Pan, einem jungen Forscher am Shanghai Artificial Intelligence Laboratory und Mitarbeitern der ShanghaiTech University und des Chinese Academy of Sciences Hangzhou Medical College,Sie entwickelten gemeinsam eine neue Vortrainingsmethode PRIME für Protein-Sequenz-Large-Language-Modelle.Gleichzeitig wurden die besten Vorhersageergebnisse bei der Vorhersage der Proteinmutationsaktivität und -stabilität sowie bei anderen temperaturbezogenen Repräsentationslernverfahren erzielt.
Die entsprechende Forschungsarbeit mit dem Titel „Ein allgemeines temperaturgesteuertes Sprachmodell zur Entwicklung von Proteinen mit verbesserter Stabilität und Aktivität“ wurde in Science Advances veröffentlicht, einer renommierten Fachzeitschrift des Science-Bereichs.
Forschungshighlights:
* PRIME kann die Leistungssteigerung bestimmter Proteinmutanten vorhersagen, ohne auf vorherige experimentelle Daten angewiesen zu sein
* PRIME kann mehrere Eigenschaften eines Proteins effektiv vorhersagen, sodass Forscher erfolgreich Proteine in unbekannten Bereichen entwickeln können
* PRIME wird auf der Grundlage eines „temperaturbewussten“ Sprachmodells trainiert, das die Temperatureigenschaften von Proteinsequenzen besser erfassen kann

Papieradresse:
https://www.science.org/doi/10.1126/sciadv.adr2641
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: 96 Millionen Datensätze zur Untersuchung der Beziehung zwischen Proteinsequenz und Temperatur
Durch die Integration öffentlicher Daten von Uniprot (Universal Protein Resource) und Proteinsequenzen, die durch Metagenomik-Studien aus Umweltproben gewonnen wurden,Forscher haben eine große Datenbank namens ProteomeAtlas zusammengestellt, die 4,7 Milliarden natürliche Proteinsequenzen enthält.
* UniProt ist eine große Datenbank, die Proteinsequenzen und zugehörige detaillierte Anmerkungen bereitstellt.
Während des Sequenz-Screening-Prozesses behielten die Forscher nur Sequenzen in voller Länge bei und verarbeiteten diese Sequenzen mit dem biologischen Sequenzalignment-Tool MMseqs2, wobei sie den Sequenzidentitätsschwellenwert auf 50% setzten, um Redundanz zu reduzieren, und dann Sequenzen identifizierten und annotierten, die mit den optimalen Wachstumstemperaturen (OGT) von Bakterienstämmen in Zusammenhang stehen.
Finale,Die Forscher haben auf diese Weise 96 Millionen Proteinsequenzen annotiert.Es bietet eine umfangreiche Ressource zur Erforschung der Beziehung zwischen Proteinsequenz und Temperatur.
Darüber hinaus wurden bei der Zero-Shot-Vorhersagefähigkeitsanalyse der thermischen Stabilität des Modells die zur Untersuchung der Schmelztemperaturänderung (ΔTm) verwendeten Datensätze aus MPTherm, FireProtDB und ProThermDB abgeleitet und alle Experimente wurden unter denselben pH-Bedingungen durchgeführt.
Unter anderem enthält MPTherm experimentelle Daten zur thermischen Stabilität von Proteinen. FireProtDB wird speziell zum Speichern experimenteller Mutationsdaten im Zusammenhang mit der thermischen Stabilität und Funktion von Proteinen verwendet. ProThermDB sammelt speziell Daten im Zusammenhang mit den thermodynamischen Eigenschaften von Proteinen. Gleichzeitig kombinierten die Forscher auch Deep Mutation Scanning (DMS)-Daten, hauptsächlich aus der Proteinmutationsanalyse-Datenbank ProteinGym.
* ProteinGym-Proteinmutationsdatensatz
https://go.hyper.ai/YlMT5
Modellarchitektur: Deep-Learning-Modell basierend auf „Temperaturwahrnehmung“
Das vom Institut vorgeschlagene neue Deep-Learning-Modell PRIME (Protein language model for Intelligent Masked pretraining and Environment prediction),Fähigkeit, Leistungsverbesserungen bestimmter Proteinmutanten vorherzusagen, ohne sich auf vorherige experimentelle Daten zu verlassen.
Das Modell wird auf der Grundlage eines „temperaturbewussten“ Sprachmodells trainiert, das auf einem Datensatz von 96 Millionen Proteinsequenzen basiert, die Aufgabe der maskierten Sprachmodellierung (MLM) auf Token-Ebene und das Vorhersageziel der optimalen Wachstumstemperatur (OGT) auf Sequenzebene kombiniert und den Korrelationsverlustterm durch Multi-Task-Lernen einführt. Es kann Proteinsequenzen mit hoher Temperaturtoleranz herausfiltern, um deren Stabilität und biologische Aktivität zu optimieren.
Speziell,PRIME besteht aus 3 Hauptteilen:Wie in der Abbildung unten gezeigt. Das erste ist das Encoder-Modul, ein Transformer-Encoder, der zum Extrahieren latenter Merkmale der Sequenz verwendet wird. Das zweite ist das MLM-Modul, das dem Encoder dabei helfen soll, die kontextuelle Darstellung von Aminosäuren zu erlernen. Gleichzeitig kann das MLM-Modul auch zum Mutant Scoring verwendet werden. Die dritte Komponente ist das OGT-Vorhersagemodul, das anhand der potenziellen Darstellung den OGT des Organismus vorhersagen kann, in dem sich das Protein befindet.
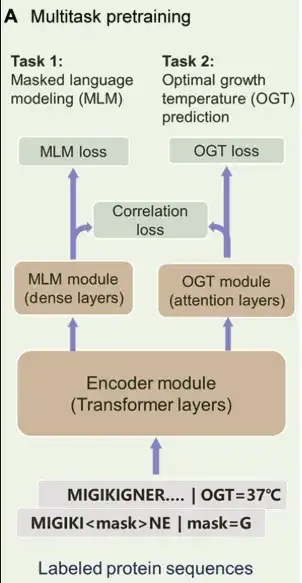
Das Multitasking-Lernen von PRIME in der Vortrainingsphase umfasst MLM, OGT-Vorhersage und Korrelationsverlust.
In,MLM wird häufig als Vortrainingsmethode für die Sequenzdatendarstellung verwendet.In dieser Studie wurden verrauschte Proteinsequenzen als Eingabe verwendet, einige Beschriftungen wurden maskiert oder durch alternative Beschriftungen dargestellt und das Trainingsziel bestand darin, diese verrauschten Beschriftungen zu rekonstruieren. Dieser Ansatz hilft dem Modell, Abhängigkeiten zwischen Aminosäuren und Kontextinformationen der Sequenz zu erfassen und gleichzeitig diesen Rekonstruktionsprozess zur Bewertung von Mutationen zu nutzen.
Die zweite Trainingsaufgabe wurde unter überwachten Bedingungen optimiert und die Forscher verwendeten einen mit OGT annotierten Datensatz von 96 Millionen Proteinsequenzen, um das PRIME-Modell zu trainieren. Die Eingabe dieser Aufgabe ist eine Proteinsequenz und die vom OGT-Modul generierten Temperaturwerte liegen zwischen 0° und 100°C. Es ist erwähnenswert, dass das OGT-Modul und das MLM-Modul mit einem gemeinsamen Encoder arbeiten.Diese Struktur ermöglicht es dem Modell, gleichzeitig Informationen zum Aminosäurekontext sowie darin enthaltene temperaturabhängige Sequenzmerkmale zu erfassen.
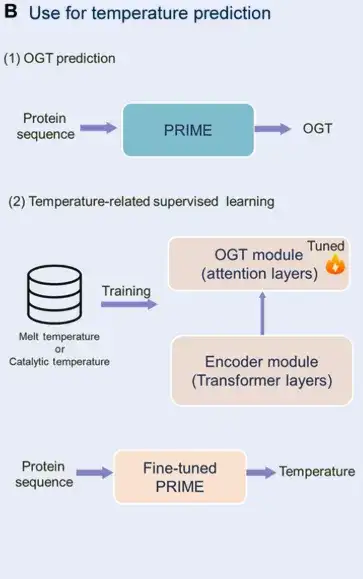
Schließlich führten die Forscher den Korrelationsverlust ein, um das Feedback vom vorhergesagten OGT zur MLM-Klassifizierung zu erleichtern und die Aufgabeninformationen auf Token- und Sequenzebene auszurichten.Dadurch kann das große Modell die Temperatureigenschaften von Proteinsequenzen besser erfassen.
Experimentelles Fazit: Übertrifft die fortschrittlichsten Methoden bei der Vorhersage der Anpassungsfähigkeit mutierter Proteinsequenzen
Die Forscher verglichen experimentell die Zero-Shot-Vorhersagefähigkeiten von PRIME mit denen der fortschrittlichsten Modelle für thermische Stabilität, darunter die Deep-Learning-Modelle ESM-1v, ESM-2, MSA-Transformer, Tranception-EVE, CARP, MIF-ST, SaProt, Stability Oracle und die traditionellen Rechenmethoden GEMME und Rosetta.
Die Forscher verwendeten Datensätze von MPTherm, FireProtDB und ProThermDB, die Schmelztemperaturänderungen (ΔTm) enthalten, die unter derselben pH-Umgebung erfasst wurden, und stellten sicher, dass für jedes Protein mindestens 10 Datenpunkte vorhanden waren, was insgesamt 66 Erkennungen ergab. Die Studie umfasste auch einen Deep Mutation Scanning (DMS)-Test, wobei ProteinGym als Prüfstand diente.
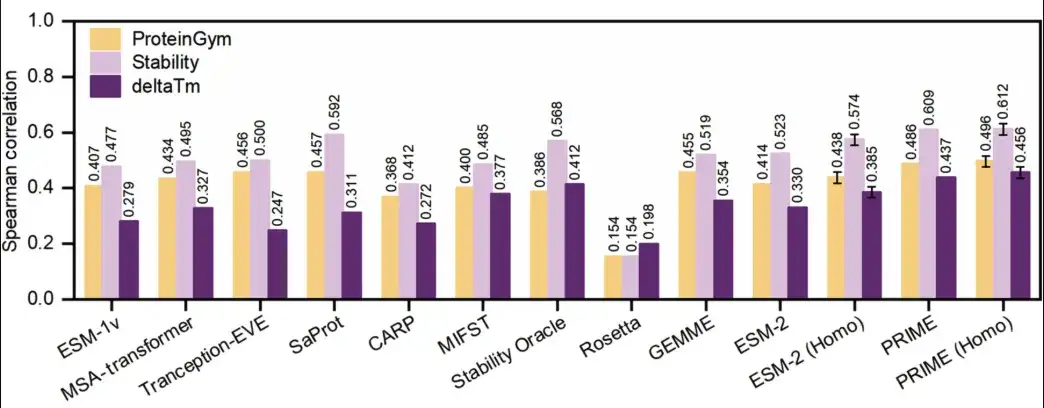
Die Ergebnisse sind in der folgenden Abbildung dargestellt.PRIME übertrifft alle anderen Methoden bei der Vorhersage sowohl der Proteinverfügbarkeit als auch der Proteinstabilität.
Im ProteinGym-Benchmark (gelb in der Abbildung unten) erreichte PRIME 0,486 und das zweitplatzierte SaProt 0,457. Im ΔTm-Datensatz (dunkelviolett in der Abbildung unten) belegt PRIME mit einer Punktzahl von 0,437 immer noch den ersten Platz und den zweiten Platz mit einer Punktzahl von 0,412. Darüber hinaus verglichen die Forscher PRIME auch mit anderen Methoden im ProteinGym-Stabilitäts-Unterdatensatz (hellviolett in der Abbildung unten), und PRIME übertraf immer noch alle anderen Methoden.
Es ist erwähnenswert, dass zur Prüfung der Wirksamkeit und Wirkung von PRIME in der praktischen Anwendung des Protein-EngineeringsDie Forscher führten außerdem ein Nassexperiment durch und wählten fünf Proteine zur Überprüfung aus.Beinhaltet LbCas12a, T7-RNA-Polymerase, Kreatinase, künstliche Nukleinsäurepolymerase und die variable Schwerkettenregion eines spezifischen Nanoantikörpers.
Bei den experimentellen Tests der 30–45 wichtigsten Einzelpunktmutationen waren mehr als 30% der von der KI empfohlenen Einzelpunktmutanten den Wildtypproteinen in Schlüsseleigenschaften wie thermischer Stabilität, enzymatischer Aktivität, Antigen-Antikörper-Bindungsaffinität, nicht-natürlicher Fähigkeit zur Nukleinsäurepolymerisation oder Toleranz unter extrem alkalischen Bedingungen deutlich überlegen, und die Positivrate einzelner Proteine übertraf 50%.
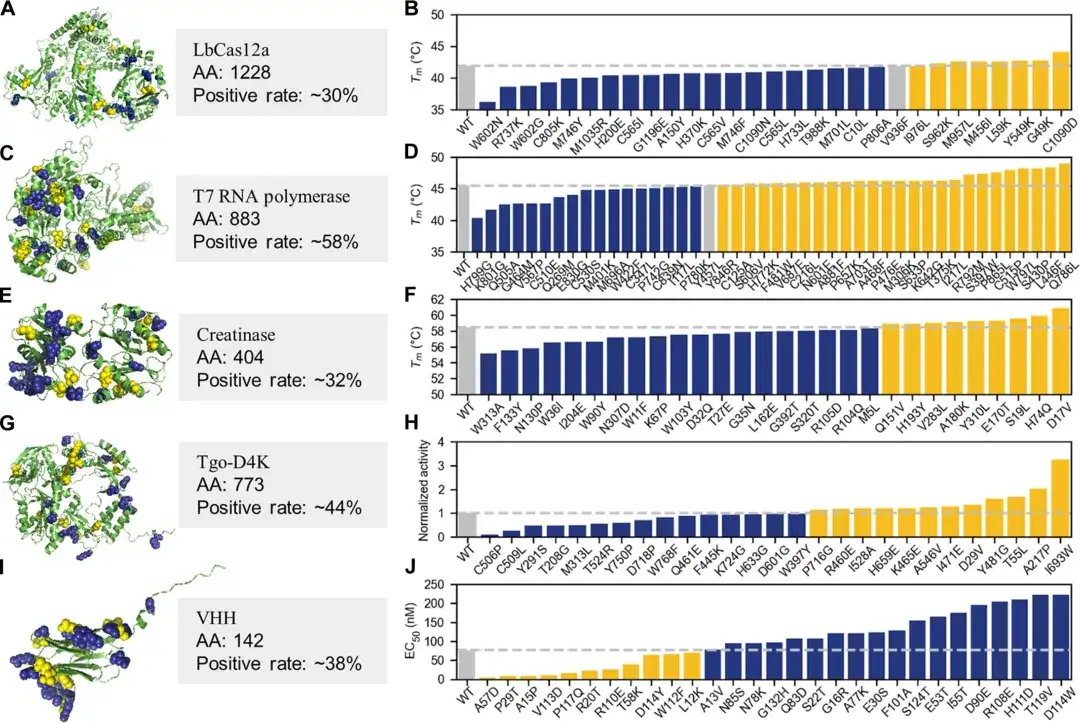
Erwähnenswert ist, dass das Team auch eine effiziente Methode auf Basis von PRIME demonstriert hat.Multi-Site-Mutanten mit erhöhter Aktivität und Stabilität können schnell erhalten werden.Durch diese Feinabstimmungsmethode mit kleinen Proben können in 2–4 Evolutionsrunden mit weniger als 100 feuchten Versuchsproben sehr gute Proteinmutanten erzeugt werden.
Beispielsweise gelang es der T7-RNA-Polymerase nach vier Runden Trocken-Nass-Iterationen, Multipunktmutanten mit hoher Aktivität und hoher Stabilität zu erzeugen. Der höchste Multipunktmutant hatte einen um 12,8 °C höheren Tm-Wert als der Wildtyp und seine Aktivität war fast viermal so hoch wie die des Wildtyps. Die Leistung einiger Produkte übertraf die ähnlicher Produkte, die den Markt seit 10 Jahren von einem international führenden Biotechnologieunternehmen (New England Biolabs) dominierten. Darüber hinaus kann Pro-PRIME in den Experimenten mit LbCas12a und T7-RNA-Polymerase negative Einzelpunktmutationen überlagern, um positive Mehrfachpunktmutationen zu erhalten.
Dies zeigt, dass PRIME die epistatischen Effekte von Proteinmutationen aus Sequenzdaten lernen kann, was für das traditionelle Protein-Engineering von großer Bedeutung ist.
Vertiefung des Protein-Engineerings zur Überwindung des Problems kleiner Proben
Im Bereich des Protein-Engineerings sind für die Proteinexpression, -reinigung und Funktionstests in der Regel teure Reagenzien und Instrumente erforderlich. Zudem sind die Experimente zeitaufwändig, was die Anzahl der erzeugbaren Proben stark einschränkt. In der Proteinfunktionsforschung erfordert das Testen der Auswirkungen von Proteinmutationen auf Funktionen (wie katalytische Aktivität, thermische Stabilität, Bindungsaffinität usw.) präzisere und komplexere Experimente, und es ist schwierig, die Leistung aller möglichen Mutationen auf einmalige Weise mit hohem Durchsatz zu messen.
Dies erschwert es maschinellen Lernmodellen, anhand begrenzter Stichproben ein ausreichendes Training zu erzielen, was zu einer schlechten Leistung des Modells bei der Vorhersage neuer Mutationen führt. Darüber hinaus können experimentelle Fehler oder Rauschen in kleinen Stichprobendaten das Modelltraining stärker beeinträchtigen. Man kann sagen, dassDie Herausforderung, nur wenige Datenproben zu sammeln, hat die Effizienz und Genauigkeit der Forschung im Bereich des Protein-Engineerings in gewissem Maße eingeschränkt.Dies hat Forscher stark dazu motiviert, innovative Technologien zu erforschen und dabei maschinelles Lernen, experimentelle Techniken und multimodale Datenanalyse zu kombinieren, um die Beschränkungen kleiner Stichproben zu überwinden.
Das in diesem Artikel beschriebene Forschungsteam hat in dieser Hinsicht hervorragende Leistungen erbracht. Zusätzlich zu dem oben erwähnten PRIME haben das Team von Professor Hong Liang und Dr. Tan Pan auch eine Reihe von Ergebnissen zum Thema Small Sample Learning veröffentlicht.
Zuvor verwendete das Team eine Kombination aus Meta-Transfer-Learning (MTL), Learning to Rank (LTR) und parametereffizienter Feinabstimmung (PEFT).Wir haben eine Trainingsstrategie namens FSFP entwickelt, mit der Proteinsprachenmodelle auch bei extremer Datenknappheit effektiv optimiert werden können.Es kann zum Lernen der Proteinanpassungsfähigkeit anhand kleiner Stichproben verwendet werden. Es verbessert die Wirkung des herkömmlichen Vortrainings großer Proteinmodelle bei der Vorhersage von Mutationseigenschaften erheblich, wenn nur sehr wenige nasse experimentelle Daten verwendet werden, und zeigt auch großes Potenzial in praktischen Anwendungen.
Die entsprechende Forschung trug den Titel „Steigerung der Effizienz von Proteinsprachenmodellen mit minimalen Labordaten durch Few-Shot-Learning“ und wurde in Nature Communications, einer Tochtergesellschaft von Nature, veröffentlicht.
Darüber hinaus hat auch Professor Hong Liang relevante Ansichten geäußert. Er ist davon überzeugt, dass „in den nächsten drei Jahren in den Bereichen Proteindesign, Arzneimittelentwicklung, Krankheitsdiagnose, Entdeckung neuer Zielmoleküle, Design chemischer Synthesewege und Materialdesign die allgemeine künstliche Intelligenz in professionellen Bereichen einen deutlichen Paradigmenwechsel herbeiführen wird. Dabei wird das wissenschaftliche Entdeckungsmodell, das sich in der Vergangenheit auf sporadisches Ausprobieren des menschlichen Gehirns stützte, in ein KI-basiertes, großformatiges, automatisiertes Standarddesignmodell umgewandelt.“
Zu den spezifischen Änderungen gehören die Entwicklung von Lernmethoden mit Nullstichproben oder kleinen Stichproben sowie die Entwicklung von Technologiemodellen für das Vortraining.In Ermangelung von Daten wird zur Vorschulung mithilfe eines physischen Simulators eine große Menge gefälschter Daten mit etwas geringerer Genauigkeit generiert und anschließend mit echten und wertvollen Daten feinabgestimmt, um das bestärkende Lernen abzuschließen.
Professor Hong betonte: „Gefälschte Daten sind Daten, die nicht aus der realen Welt stammen, aber ein gewisses Maß an Zuverlässigkeit aufweisen. Sie können durch KI generiert oder durch physikalische Computersimulation zur Datenverbesserung gewonnen werden. Schließlich sind reale Nassversuchsdaten am wertvollsten und werden für die endgültige Feinabstimmung des Modells verwendet.“
Tatsächlich besteht die Herausforderung der Datenknappheit nicht nur im Bereich des Protein-Engineerings. Von entscheidender Bedeutung sind Lernmethoden mit kleinen Stichproben oder sogar Nullstichproben. Wir freuen uns darauf, dass das Team von Professor Hong Liang und Dr. Tan Pan weitere hochwertige Ergebnisse zu diesem Problempunkt erzielen wird.