Command Palette
Search for a command to run...
Im Nature Journal Veröffentlicht! Die Central China Normal University Hat DigFrag Vorgeschlagen, Das Mithilfe Künstlicher Intelligenz Molekülfragmente Präzise Segmentiert Und 44 Arzneimittel-/Pestizidmoleküle generiert.

In den letzten Jahrzehnten hat die fragmentbasierte Arzneimittelforschung (FBDD) eine wichtige Rolle in der Erforschung und Entwicklung neuer Arzneimittel gespielt, indem sie kleine Molekülfragmente identifizierte, die nur schwach mit Zielproteinen interagieren, und die Strukturinformationen dieser Fragmente optimierte, um aktivere Leitsubstanzen zu entwickeln.
Obwohl FBDD eine Schlüsselrolle bei der Entdeckung und Entwicklung von Medikamenten spielt, war die Konstruktion und das Screening effektiver Molekülfragmentbibliotheken in diesem Bereich schon immer eine große Herausforderung. Traditionelle FBDD-Methoden basieren auf empirischer Intuition, was ihre Fähigkeit zur Entwicklung vielfältiger Strukturen einschränkt. Glücklicherweise bietet die Entstehung künstlicher Intelligenz eine transformative Lösung für diese Herausforderung.
Vor Kurzem hat das Team von Professor Yang Guangfu und Associate Professor Wang Fan von der Central China Normal University eine digitale Segmentierungsmethode namens DigFrag entwickelt.Die Methode konzentriert sich lokal auf den Molekülgraphen, hebt wichtige Unterstrukturen hervor und unterteilt diese Unterstrukturen in Fragmente. Experimentelle Ergebnisse zeigen, dass die von DigFrag segmentierten Fragmente eine höhere strukturelle Vielfalt aufweisen und die auf der Grundlage dieser Fragmente erzeugten Verbindungen eher mit den erwarteten chemischen Eigenschaften übereinstimmen. Dies deutet darauf hin, dass mit KI-Methoden generierte Daten möglicherweise besser für das Training und die Anwendung von KI-Modellen geeignet sind.
Die Forschungsarbeit mit dem Titel „DigFrag als digitale Fragmentierungsmethode für die Arzneimittelentwicklung auf Basis künstlicher Intelligenz“ wurde in der internationalen Fachzeitschrift Nature Communications Chemistry veröffentlicht.
Forschungshighlights:
* Die Studie ergab, dass DigFrag-basierte Fragmente, wenn sie mit KI-Modellen kombiniert werden, effektiv Moleküle mit gewünschten Eigenschaften erzeugen können
* Durch präzises Screening identifizierte die Studie schließlich 24 Arzneimittelmoleküle und 20 Pestizidmoleküle
* Das Team entwickelte eine benutzerfreundliche Plattform namens MolFrag, die mehrere Fragmentierungstechnologien integriert, um ein breiteres Spektrum an molekularen Analyse- und Designarbeiten zu unterstützen
Papieradresse:
https://doi.org/10.1038/s42004-024-01346-5
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: selbst erstellte Datenbank PADFrag, die fast 3.000 Arten von Arzneimitteldaten enthält
Der in dieser Studie verwendete Modellierungsdatensatz stammt hauptsächlich aus der selbst erstellten Datenbank PADFrag. Konkret umfasst die PADFrag-Datenbank hauptsächlich den von der FDA zugelassenen Arzneimittelkatalog in der DrugBank-Datenbank, der 1.652 Arzneimittel enthält, und die von Alan Wood aufgelisteten kommerziellen Pestizide, insgesamt 1.259.
*PADFrag, eine Datenbank zur Erforschung bioaktiver Fragmente für die Arzneimittelforschung
https://pubs.acs.org/doi/10.1021/acs.jcim.8b00285
Um die Konsistenz und Zuverlässigkeit der Daten sicherzustellen, schloss das Forschungsteam Verbindungen mit nicht standardmäßigen Strukturen aus. Anschließend wurde der gesamte Datensatz im Verhältnis 8:1:1 in Trainingssatz, Validierungssatz und Testsatz aufgeteilt, um das Trainieren, Auswerten und Testen des Modells zu erleichtern.
DigFrag: Ein 3-stufiger Workflow zum Erhalt von Fragmenten mit größerer struktureller Vielfalt
DigFrag ist eine innovative digitale Segmentierungsmethode, die einen Graph-Attention-Mechanismus verwendet, um Arzneimittel-/Pestizidfragmente zu identifizieren und zu segmentieren. Sein Hauptvorteil besteht darin, dass es aus der Perspektive der maschinellen Intelligenz Fragmente mit höherer struktureller Vielfalt erhalten kann, anstatt sich ausschließlich auf menschliches Fachwissen zu verlassen.
Darüber hinaus wurden in die Studie die mit vier Methoden – BRICS, RECAP, MacFrag und DigFrag – segmentierten Fragmente integriert und in das DeepFMPO-Modell-Framework integriert, um Arzneimittelmoleküle zu generieren und ihre Leistung anhand verschiedener Indikatoren zu bewerten.
Schließlich entwickelten die Forscher auf der Grundlage mehrerer Technologien zur Molekülfragmentierung eine benutzerfreundliche Plattform namens MolFrag zur Unterstützung der Molekülsegmentierungsarbeit.
Konkret gliedert sich der Arbeitsablauf dieser Studie in drei Teile:
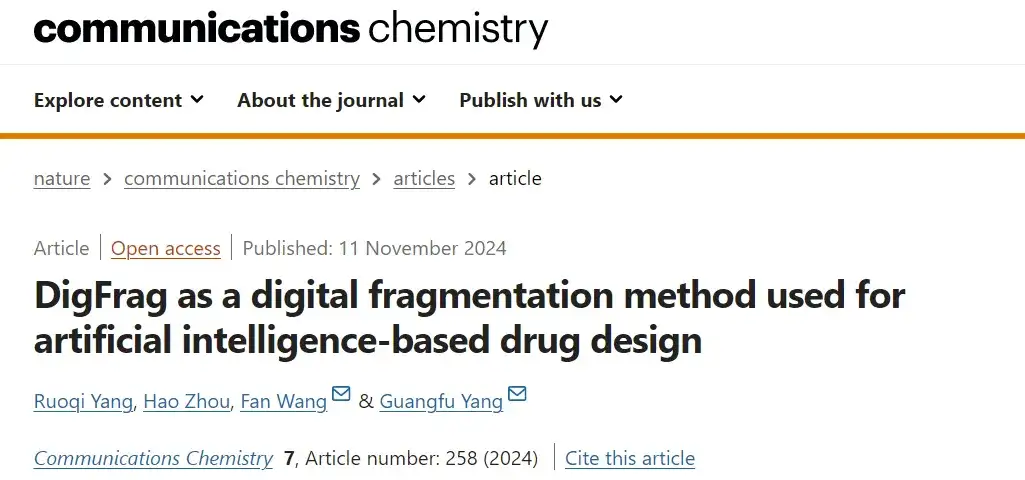
Erstens der KI-basierte Fragmentierungsansatz:Diese Studie basiert auf der Graph Neural Network (GNN)-Architektur und verwendet die DigFrag-Methode zum Fragmentieren von Molekülen.
Wie in Abbildung A oben gezeigt, definierten die Forscher den Molekülgraphen als G=(V, E), wobei V Knoten darstellt, die den Atomen im Molekül entsprechen, und E Verbindungskanten darstellt, die den chemischen Bindungen zwischen Atomen entsprechen. In diesem Prozess wird basierend auf dem Merkmalsextraktionsnetzwerk (Merkmalsmatrix) des Graph-Aufmerksamkeitsmechanismus der ursprüngliche Molekülgraph zunächst in eine Reihe von Aufmerksamkeitsebenen eingegeben, um für jedes Atom eine separate Einbettungsdarstellung zu erhalten. Diese atomaren Einbettungen werden dann zu einem einheitlichen Vektor aggregiert, der auch als Superknoten bezeichnet wird. Schließlich wird durch weitere Verarbeitung der Aufmerksamkeitsschicht die eingebettete Darstellung des gesamten Fragments erhalten.
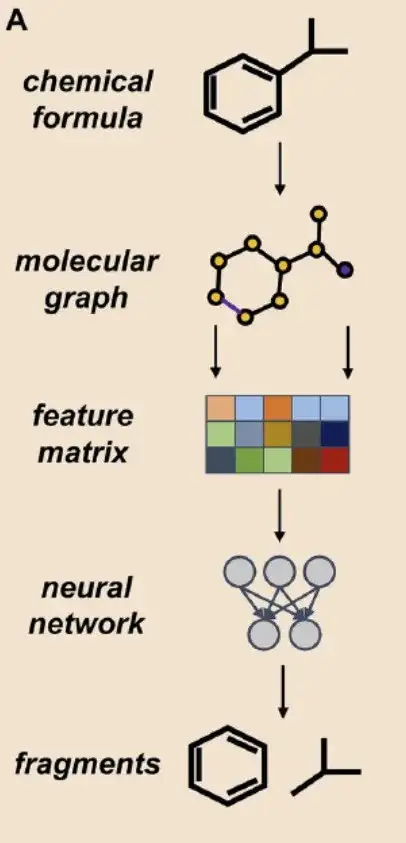
Zweitens das Actor-Critic-Modell-FrameworkUm die Auswirkungen der digitalen Segmentierung auf fragmentbasierte tiefe generative Modelle weiter zu verdeutlichen, haben die Forscher, wie in Abbildung B unten dargestellt, mit vier Methoden segmentierte Fragmente integriert: BRICS, RECAP, MacFrag und DigFrag. Außerdem haben sie für ihre Forschung die DeepFMPO-Architektur verwendet, ein Open-Source-Tool zur zweidimensionalen Molekülgenerierung auf Basis von fragmentbasiertem Verstärkungslernen.
*DeepFMPO ist ein Actor-Critic-Reinforcement-Learning-Modell, das die gewünschte Verbindung durch Ersetzen von Fragmenten in der Verbindung erhält.
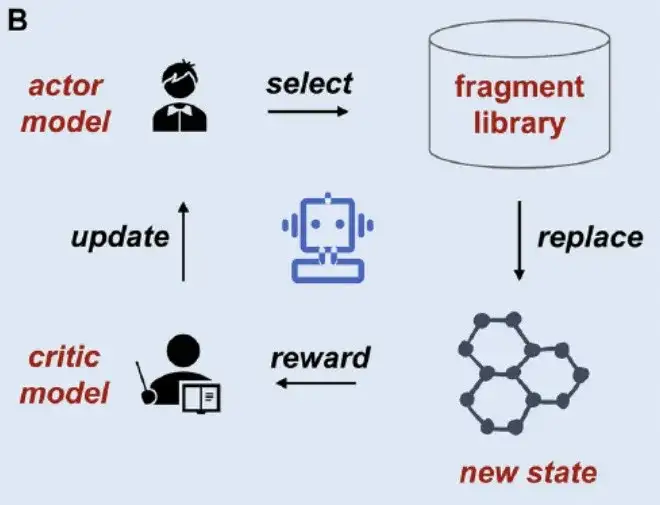
Drittens: Richten Sie eine Online-Plattform ein:Obwohl eine Vielzahl von Methoden zur Molekülfragmentierung entwickelt wurden, mangelt es an einfach zu bedienenden Online-Servern. Daher wurde im Rahmen dieser Studie, wie in Abbildung C oben dargestellt, eine benutzerfreundliche Plattform namens MolFrag entwickelt, die auf verschiedenen Fragmentierungstechniken basiert. Die Plattform kombiniert nahtlos vier Methoden zur Molekülfragmentierung: BRICS, RECAP, MacFrag und DigFrag und stellt sicher, dass sie von Forschern mit unterschiedlichem Fachwissen genutzt werden kann.
Adresse der MolFrag-Plattform:
https://dpai.ccnu.edu.cn/MolFrag
Forschungsergebnisse: Segmentierte Molekülfragmente von DigFrag weisen eine höhere Diversität auf
DigFrag-Fragmente haben eine große Anzahl drehbarer Bindungen
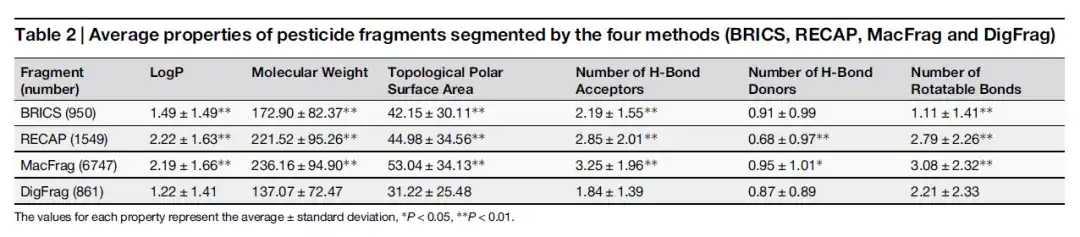
Im Rahmen der Studie wurde das Modell zunächst darauf trainiert, Arzneimittel- und Pestizidfragmente genau zu segmentieren. Anschließend führten die Forscher einen detaillierten Vergleich der drei wichtigsten Leistungsindikatoren von DigFrag durch: Modellgenauigkeit, Fläche unter der Kurve (AUC) und Matthews-Korrelationskoeffizient (MCC) der mit herkömmlichen (RECAP, BRICS) und den neuesten (MacFrag) Methoden erhaltenen Fragmente durch fünffache Kreuzvalidierung. Wie aus der folgenden Tabelle hervorgeht, ist die Verteilung der Eigenschaften der Arzneimittelfragmente bei den von DigFrag und den von BRICS segmentierten Fragmenten ähnlicher.
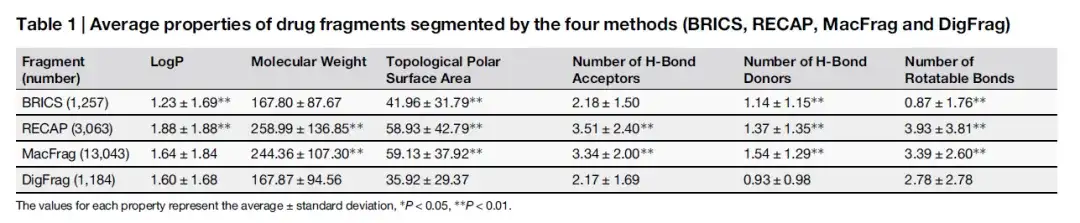
Wie aus der folgenden Tabelle hervorgeht, ähneln Molekulargewicht und Anzahl der H-Brücken-Akzeptoren der von DigFrag segmentierten Arzneimittelfragmente zwar denen der von BRICS segmentierten, die Anzahl der drehbaren Bindungen ist jedoch größer, was möglicherweise mit der einzigartigen Art des Bruchs der Ringstruktur zusammenhängt. Was Pestizidfragmente betrifft, ist das durchschnittliche Molekulargewicht der von DigFrag segmentierten Fragmente niedriger.
Segmentierte DigFrag-Fragmente weisen eine höhere strukturelle Vielfalt auf
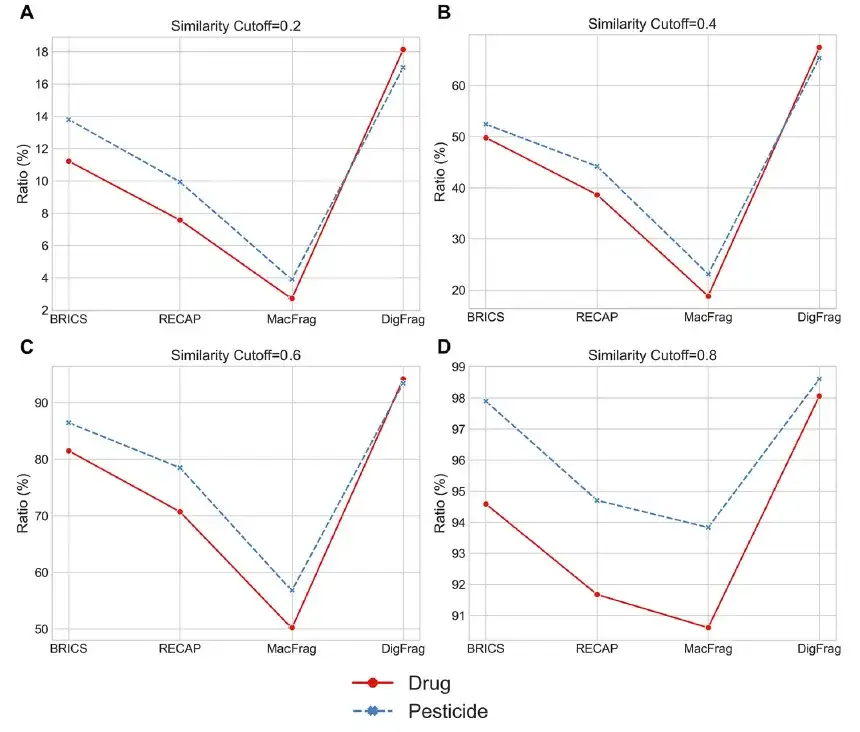
Der Schwerpunkt dieser Studie lag auf der Bewertung der strukturellen Vielfalt segmentierter Fragmente und einem Vergleich der DigFrag-Methode mit herkömmlichen Methoden (RECAP und BRICS) sowie einer hochmodernen Methode (MacFrag). Die Ergebnisse zeigten, dass die von DigFrag in Arzneimittel und Pestizide segmentierten Fragmente eine niedrigere Wiederholungsrate aufwiesen als die anderen drei Methoden, nämlich 9,97%-21,37% bzw. 8,94%-15,20%, was darauf hindeutet, dass einzigartige Fragmente erzeugt werden können. MacFrag deckt die meisten Fragmente von BRICS und RECAP ab, was darauf schließen lässt, dass es sich nicht um eine völlige Innovation handelt, sondern um eine Erweiterung traditioneller Ansätze.

Die Forscher verwendeten außerdem den t-SNE-Algorithmus, um die chemische Raumverteilung zu visualisieren. Wie in der folgenden Abbildung gezeigt, schneidet DigFrag hinsichtlich des Fragmentclusterverhältnisses gut ab, insbesondere wenn die Ähnlichkeitsschwellenwerte bei 0,4 und 0,6 liegen, was auf eine höhere strukturelle Vielfalt hinweist.
Auf DigFrag basierende Modelle erzeugen Moleküle höherer Qualität
Auf der MOSES-Benchmark-Plattform wurde in dieser Studie die Leistung verschiedener generativer Modelle verglichen. Wie aus den beiden folgenden Tabellen hervorgeht, erreichte das auf DigFrag basierende Modell einen Filter-Score von 0,828 und weist damit eine höhere Sicherheit auf, was auf die umfassende Berücksichtigung von Toxizität und Stabilität im Fragmentierungsprozess des Deep Learning zurückzuführen sein könnte.
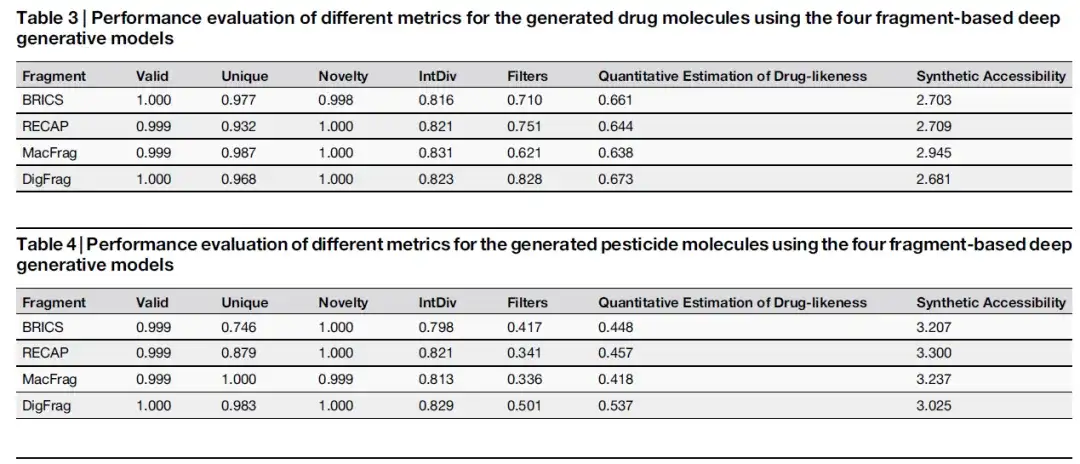
Wie in der folgenden Abbildung gezeigt, schnitten die vom DigFrag-basierten Modell generierten Molekülfragmente im Hinblick auf Pestizidmoleküle hinsichtlich SMILES-Validität, Neuheit, Skelettdiversität und Strukturwarnungen gut ab. Darüber hinaus übertrafen die vom DigFrag-Modell generierten Molekülfragmente von Arzneimitteln und Pestiziden andere Modelle bei der Mittelwertanalyse der quantitativen Schätzung (QED) und der synthetischen Zugänglichkeit (SA).
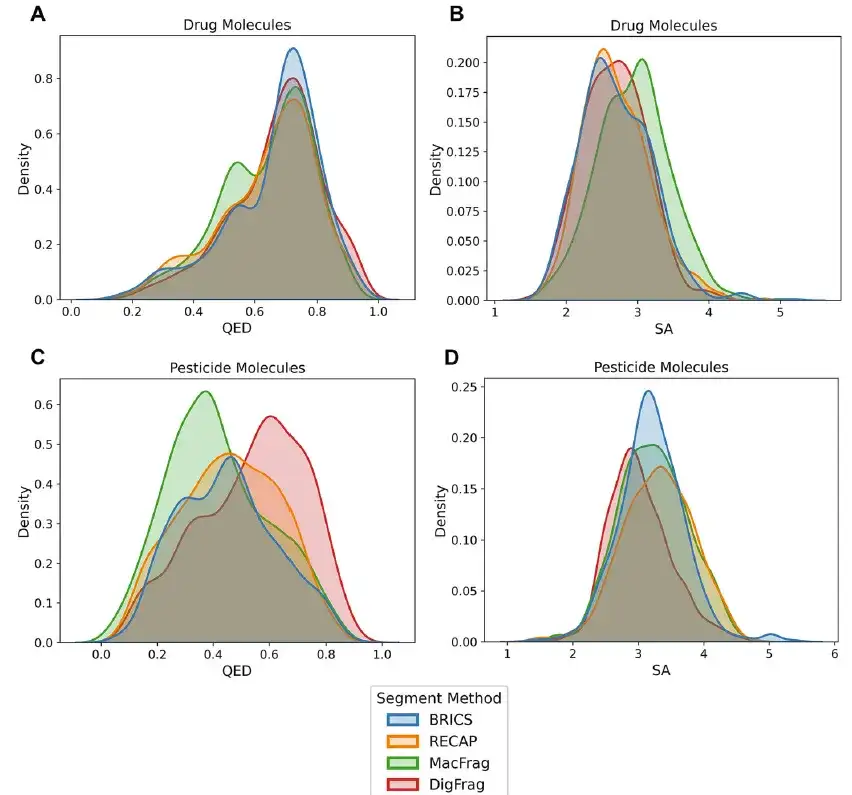
Darüber hinaus weisen die von DigFrag segmentierten Molekülfragmente hinsichtlich Molekulargewicht, QED und SA-Eigenschaftsverteilung die größte Ähnlichkeit mit dem MOSES-Datensatz auf. Diese Ergebnisse deuten darauf hin, dass das DigFrag-Modell Moleküle höherer Qualität produzieren kann, und unterstreichen gleichzeitig die Präferenz des KI-Modells für KI-abgeleitete Daten im Moleküldesign, was die Anwendungsvorteile der KI-Technologie in diesem Bereich hervorhebt.
Ausgewählte 44 hocheffiziente und energiearme Arzneimittel- und Pestizidmoleküle
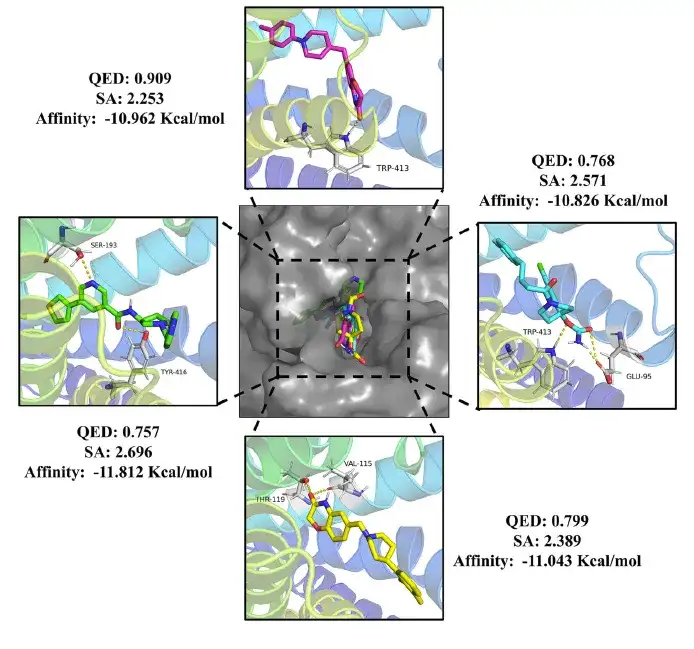
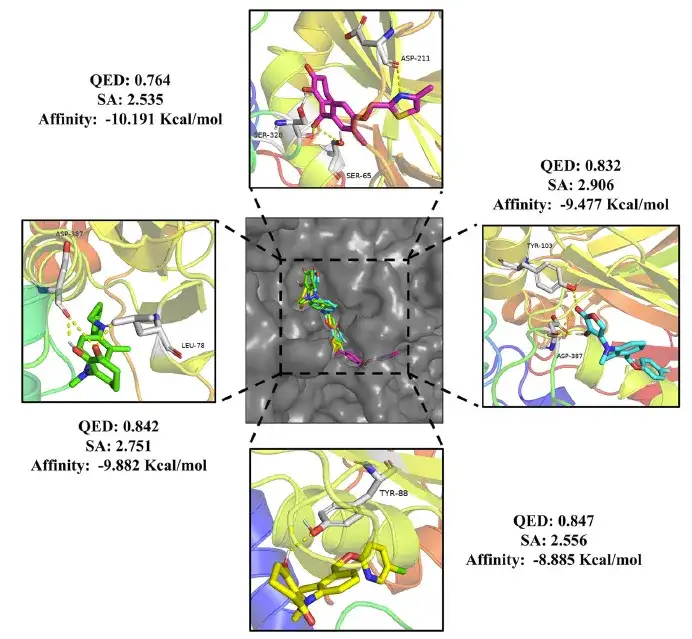
Schließlich wurden im Rahmen der Studie nach einer genauen Prüfung 24 Arzneimittelmoleküle und 20 Pestizidmoleküle identifiziert, die alle die Kriterien von QED-Werten über 0,75, SA-Werten unter 3 und einer geringeren freien Bindungsenergie als Domperidon (-10,7 Kcal/mol) und Methotriazin (-8,4 Kcal/mol) erfüllten.
Die Studie analysierte außerdem die Wechselwirkungen dieser Moleküle mit ihren Zielen. Wie in der Abbildung unten gezeigt, ergab die Studie, dass das Arzneimittelmolekül effektiv an die aktive Tasche von DRD2 binden und Wasserstoffbrücken mit wichtigen Aminosäureresten bilden kann.
Darüber hinaus sind die Pestizidmoleküle, wie in der Abbildung unten gezeigt, durch die Bildung von Wasserstoffbrücken stabil an die Aminosäurereste von HPPD gebunden. Im Vergleich zu den positiven Arzneimitteln zeigten die erzeugten Verbindungen auch unterschiedliche Bindungsarten, was auf die Möglichkeit unterschiedlicher pharmakologischer Mechanismen schließen lässt und neue Richtungen für die zukünftige Forschung liefert.
Der Einsatz von KI in der Arzneimittelforschung verändert die Spielregeln
Derzeit wird die Anwendung von KI in der Arzneimittelforschung immer umfassender. Durch Deep-Learning-Netzwerke sind KI-Modelle in der Lage, komplexe biologische Daten und chemische Strukturen zu analysieren, um die Aktivität und Selektivität von Arzneimittelmolekülen vorherzusagen.
Das in dieser Studie erwähnte Team von Professor Yang Guangfu und Associate Professor Wang Fan hat Anfang des Jahres außerdem gemeinsam ein multimodales Deep-Learning-Architekturmodell namens Pesti-DGI-Net zur Vorhersage pestizidähnlicher Eigenschaften entwickelt. Es kann die pestizidähnlichen Eigenschaften von Verbindungen vorhersagen, indem es drei molekulare Darstellungsformen integriert: molekulare Deskriptoren, molekulare Bilder und molekulare Graphen. Die Ergebnisse zeigen, dass Pesti-DGI-Net bei mehreren Indikatoren eine überlegene Leistung aufweist.
Link zum Artikel:
https://doi.org/10.1016/j.compag.2024.108660
Darüber hinaus wurden mithilfe künstlicher Intelligenz in jüngster Zeit auch im Bereich der Arzneimittelforschung fruchtbare Ergebnisse erzielt. Vor nicht allzu langer Zeit hat das Shanghai Institute of Nutrition and Health der Chinesischen Akademie der Wissenschaften ein Dual-View-Deep-Learning-Modell namens JointSyn entwickelt, um die synergistischen Effekte von Medikamentenkombinationen vorherzusagen. Die Ergebnisse zeigen, dass JointSyn hinsichtlich Vorhersagegenauigkeit und Robustheit bei verschiedenen Benchmarks die aktuellen modernsten Methoden übertrifft.
Link zum Artikel:
https://doi.org/10.1093/bioinformatics/btae604
Neben ihrer Anwendung bei der Vorhersage von Arzneimitteleigenschaften hat die KI-Technologie auch in vielen anderen Bereichen bemerkenswerte Forschungsergebnisse erzielt, beispielsweise bei der Optimierung des Arzneimitteldesigns, der toxikologischen und Sicherheitsbewertung, der Gestaltung klinischer Studien und der Patientenauswahl. Es ist absehbar, dass der Einsatz von KI in der Arzneimittelforschung die Spielregeln der Arzneimittelentwicklung neu gestaltet. Durch die kontinuierliche Weiterentwicklung der Technologie können Patienten sicherere und wirksamere Behandlungsmöglichkeiten geboten werden, indem die Genauigkeit von Vorhersagen verbessert, das Arzneimitteldesign optimiert und Entwicklungskosten und -zeit reduziert werden.








