Command Palette
Search for a command to run...
Realisieren Sie Eine Dynamische Protein-Docking-Vorhersage! Die Shanghai Jiao Tong University/Xingyao Technology/Sun Yat-sen University Und Andere Haben Gemeinsam Das Geometrische Tiefengenerierungsmodell DynamicBind Auf Den Markt Gebracht

Eiweiß ist die materielle Grundlage des Lebens. Seine Funktion ist eng mit der Dynamik der Proteinstruktur und -konformation verbunden und wird durch Liganden reguliert. Die Untersuchung von Protein-Liganden-Interaktionen ist für die Arzneimittelentdeckung und das Screening von großer Bedeutung. Rückblickend auf den Forschungsfortschritt ist die Einführung von AlphaFold ein bahnbrechender Durchbruch, der die räumliche dreidimensionale Struktur eines einzelnen Proteins vorhersagen und eine strukturelle Grundlage für die Untersuchung von Protein-Liganden-Interaktionen bieten kann.
AlphaFold kann jedoch nur die statische Struktur eines Proteins zu einem bestimmten Zeitpunkt vorhersagen und versagt bei der Vorhersage dynamischer Veränderungen der Proteinstruktur.Wenn die von AlphaFold vorhergesagte ligandenfreie Proteinstruktur als Eingabe für das Docking verwendet wird, stimmen die resultierenden Vorhersagen der Ligandenposition häufig nicht mit der ligandengebundenen Cokristallstruktur überein. Darüber hinaus ist es unwahrscheinlich, dass die von AlphaFold vorhergesagte Struktur die günstigsten Seitenketten- und Hauptkettenkonfigurationen für die Ligandenbindung aufweist, was dazu führt, dass sich die relevanten aktiven Stellen nicht an der richtigen Position befinden. Daher ist es derzeit schwierig, die AlphaFold-Struktur für das Screening und die Entwicklung von Medikamenten zu verwenden.
In Anbetracht dessenDie von Zheng Shuangjia von der Shanghai Jiao Tong University geleitete Forschungsgruppe hat in Zusammenarbeit mit Star Pharma Technology, der Sun Yat-sen University School of Pharmacy und der Rice University ein geometrisches, tiefes generatives Modell namens DynamicBind vorgeschlagen, das für das dynamische Docking von Proteinen entwickelt wurde.Es kann die Proteinkonformation effektiv vom anfänglichen, durch AlphaFold vorhergesagten Zustand in einen holoähnlichen Zustand anpassen und so ein neues Forschungsparadigma auf der Grundlage von Deep Learning bereitstellen und die dynamischen Veränderungen von Proteinen für die Arzneimittelentwicklung in der Post-AlphaFold-Ära berücksichtigen.Diese Methode wurde auch durch Nassexperimente im internationalen Arzneimittel-Screening-Wettbewerb CACHE verifiziert und kann wettbewerbsfähige Leitsubstanzen für schwer medikamentös zu behandelnde Ziele zur Behandlung der Parkinson-Krankheit screenen.
Die Studie mit dem Titel „DynamicBind: Vorhersage der ligandenspezifischen Protein-Ligand-Komplexstruktur mit einem tiefen äquivarianten generativen Modell“ wurde in Nature Communications veröffentlicht.
Forschungshighlights:
* Durch den Einsatz fortschrittlicher Deep-Diffusion-Modelle und neuronaler Netzwerktechnologie mit äquivarianter Geometrie werden die Generierung der Proteinkonformation und die Vorhersage der Ligandenpose in einem Rahmen vereint, wodurch eine dynamische Docking-Vorhersage von Proteinen und Liganden ermöglicht wird
* DynamicBind übertrifft herkömmliche Docking-Methoden und Deep-Learning-basierte starre Docking-Methoden beim Protein-Liganden-Docking
* DynamicBind nutzt die von AlphaFold vorhergesagte Proteinkonformation, um die Proteinkonformation dynamisch anzupassen und die optimale Konformation zu finden, die am besten zum Liganden passt

Papieradresse:
https://www.nature.com/articles/s41467-024-45461-2
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Dataset: Basierend auf dem PDBbind-Dataset wird das MDT-Testset verwendet, um den Evaluierungsumfang zu erweitern
Die Forscher verwendeten zunächst den PDBbind-Datensatz, um die Trainings-, Validierungs- und Testsätze des Modells in chronologischer Reihenfolge zu trainieren, gepaart mit experimentell gemessenen Bindungsaffinitäten.Da der PDBbind-Testsatz etwa 300 Strukturen aus dem Jahr 2019 enthält, darunter viele Liganden, die keine kleinen Moleküle sind (53 sind Peptide), erweiterten die Forscher den Umfang der Bewertung mit einem kuratierten Testsatz für wichtige Arzneimittelziele (MDT).
Der MDT-Testsatz umfasst 599 Strukturen, die im Jahr 2020 oder später archiviert wurden, darunter medikamentenähnliche Liganden und Proteine aus vier Hauptfamilien: Kinasen, GPCRs, Kernrezeptoren und Ionenkanäle. Diese Proteinfamilien stellen die Ziele von etwa 701 von der FDA zugelassenen TP3T-basierten niedermolekularen Arzneimitteln dar, was repräsentativ ist.
DynamicBind: Ein geometrisches Deep-Learning-basiertes Modell zur Vorhersage der Struktur dynamischer Komplexe
Im Unterschied zu herkömmlichen Docking-Methoden, die Proteine als überwiegend starre Einheiten behandeln, verwendet DynamicBind fortschrittliche Deep-Diffusion-Modelle und neuronale Netzwerktechnologie mit äquivarianter Geometrie, um die beiden traditionell getrennten Schritte der Proteinkonformationserzeugung und der Ligandenpose-Vorhersage in einem einzigen Rahmen zu vereinen und so eine dynamische Docking-Vorhersage von Proteinen und Liganden zu erreichen.Gleichzeitig ist es als durchgängige Deep-Learning-Methode bei der Erfassung einer breiten Palette von Proteinkonformationsänderungen um mehrere Größenordnungen schneller als herkömmliche MD-Simulationen.
DynamicBind akzeptiert PDB-formatierte apo-ähnliche Strukturen und mehrere weit verbreitete Ligandenformate für kleine Moleküle, wie SMILES oder SDF.Während der Inferenz platziert das Modell zufällig Liganden und mithilfe von RDKit werden Keimkonformationen der Liganden um das Protein herum generiert. Während der Trainingsphase zielt das Modell darauf ab, den Prozess von der Apo-ähnlichen Konformation zur Holo-Konformation zu erlernen. Während der Inferenz iteriert das Modell 20 Mal auf der ursprünglichen Eingabestruktur.
Wie in Abbildung a unten gezeigt, stellt Pink den holografischen Zustand (Holo) der Proteinkonformation dar, Grün das ursprüngliche Apolipoprotein und die vom Modell vorhergesagten Konformationen, Cyan den nativen Liganden und Orange den vorhergesagten Liganden.
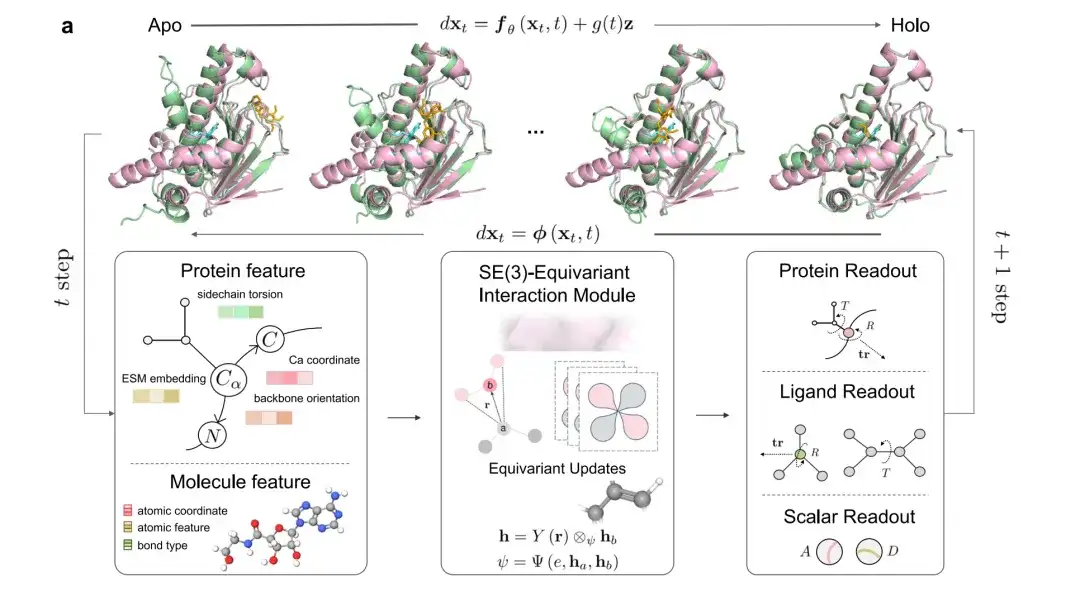
Bei jeder Iteration werden die Protein- und Ligandenmerkmale und -koordinaten (einschließlich Seitenkettentorsion, Ca-Atomkoordinaten usw.) in ein SE(3) Equivariantes Interaktionsmodul eingegeben. Die Ergebnisse der Modellausgabe umfassen die globale Translation und Rotation des Liganden und jedes Proteinrests, den Torsionswinkel des Liganden und die Rotation des Chi-Winkels der Proteinreste sowie zwei Vorhersagemodule (Bindungsaffinität A und Vertrauenswert D).
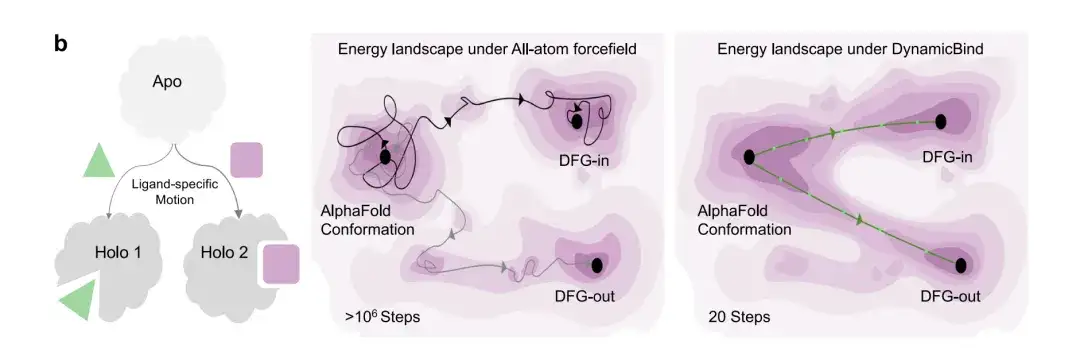
Abbildung b unten zeigt die Sampling-Effizienz des DynamicBind-Modells. Während des Übergangs des Kinaseproteins von DFG-in zu DFG-out kann das Modell zwei verschiedene Holo-Konformationen vorhersagen, wenn das Protein an zwei verschiedene Liganden bindet. Darüber hinaus kann DynamicBind die Konformation des gebundenen Proteins innerhalb von 20 Schritten vorhersagen, während MD-Simulationen auf Atomebene Millionen von Schritten erfordern, um denselben Bindungszustand zu finden.
DynamicBind ist ein Allrounder in der Vorhersage dynamischer Protein-Dockingvorgänge. Es erfüllt fünf Hauptaufgaben gut.
Um die Leistung des DynamicBind-Modells zu bewerten, testeten die Forscher es anhand von fünf Aufgaben, darunter:
(1) Vergleichen Sie DynamicBind mit aktuellen Dockingmethoden.
(2) die Fähigkeit, Konformationsänderungen in einer großen Anzahl von Proteinen zu untersuchen;
(3) der Umfang der Behandlung von Proteinkonformationsänderungen;
(4) die Fähigkeit, versteckte Taschen vorherzusagen, um ein dynamisches Andocken zu erreichen;
(5) Screeningleistung bei Antibiotika-Benchmarktests.
DynamicBind übertrifft herkömmliche Docking-Methoden und Deep-Learning-basierte starre Docking-Methoden
Während des Tests verwendeten die Forscher keine holografischen Strukturen als Eingabe und gingen davon aus, dass keine holografischen Proteinkonformationen verfügbar waren. Sie verwendeten als Eingabe nur die von AlphaFold vorhergesagten Proteinkonformationen.Da die Holokonformationen eine starke Form- und Ladungskomplementarität mit den kokristallisierten Liganden aufweisen, wird der Prozess der Ligandenpose-Vorhersage vereinfacht.
Wie in den Abbildungen a und b unten gezeigt, verglichen die Forscher DynamicBind mit anderen Basismodellen im PDBbind-Datensatz und im MDT-Datensatz. Unter verschiedenen RMSD-Schwellenwerten übertraf DynamicBind andere Methoden. Insbesondere beträgt der Anteil der DynamicBind-Liganden mit RMSD-Schwellenwerten unter 2 Å (5 Å) 331 TP3T (651 TP3T) im PDBbind-Testsatz und 391 TP3T (681 TP3T) im MDT-Testsatz.
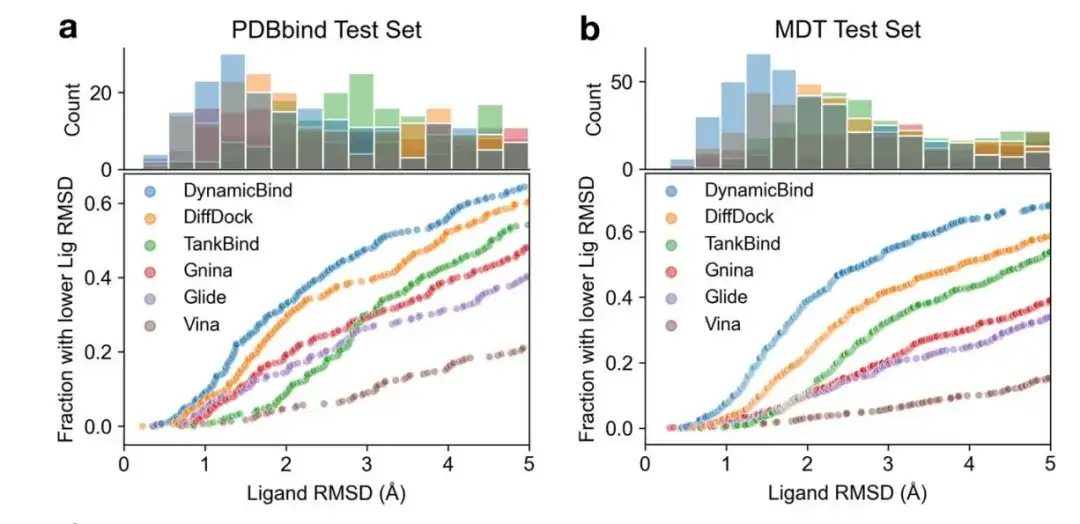
Bei der Auswertung des Modells wird jedoch nur der RMSD-Wert des Liganden zur Bewertung verwendet, was für Deep-Learning-basierte Modelle wie DiffDock, TankBind und DynamicBind von Vorteil ist, da sie eine höhere Toleranz gegenüber Konformationskonflikten aufweisen. Es ist jedoch ungünstig für die Dockingmethoden Gnina, Glide und Vina, die strikt Van-der-Waals-Kräfte implementieren und auf Kraftfeldern basieren, wodurch die Objektivität der Modellbewertung beeinträchtigt wird. Daher verwendeten die Forscher Liganden-RMSD- und Konfliktwerte, um die Erfolgsrate der Ligandenvorhersage zu bewerten.
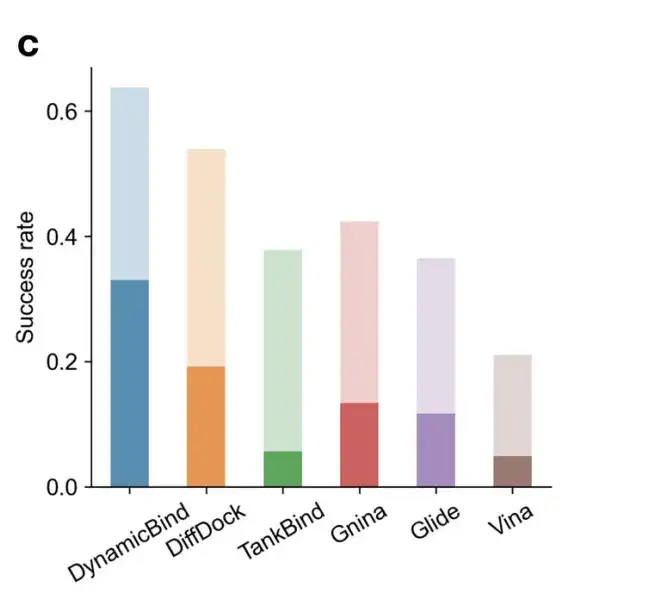
Abbildung c zeigt die Erfolgsrate der Ligandenvorhersage unter Verwendung strenger Kriterien (Ligand-RMSD < 2 Å, Konflikt-Score < 0,35) und lockererer Kriterien (Ligand-RMSD < 5 Å, Konflikt-Score < 0,5). Unter strengeren Bedingungen ist die Erfolgsrate von DynamicBind (0,33) 1,7-mal so hoch wie die des besten Basis-DiffDock (0,19).
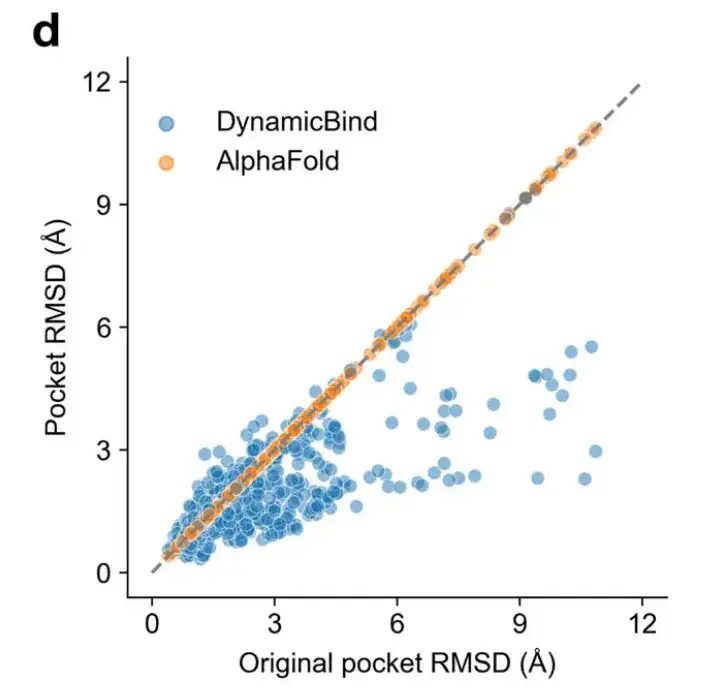
Darüber hinaus ist der von DynamicBind vorhergesagte Taschen-RMSD, selbst wenn der RMSD zwischen der ursprünglichen Tasche und der Kristallstruktur groß ist, deutlich kleiner als der von AlphaFold vorhergesagte, wie in Abbildung d unten gezeigt.
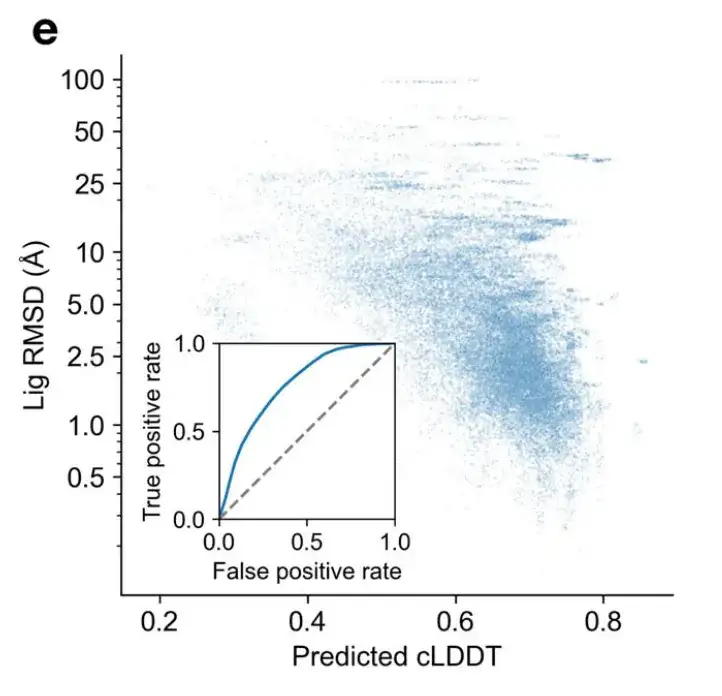
Angesichts der Fähigkeit von DynamicBind, verschiedene Konformationen zu erzeugen, und inspiriert von den LDDT-Werten von AlphaFold entwickelten die Forscher ein Kontakt-LDDT-Bewertungsmodul (cLDDT), um aus der vorhergesagten Ausgabe die am besten geeignete komplexe Struktur auszuwählen.
Wie in Abbildung e unten gezeigt, weist der von DynamicBind vorhergesagte cLDDT eine gute Korrelation mit dem tatsächlichen RMSD des Liganden auf, was auf seine Wirksamkeit bei der Auswahl hochwertiger komplexer Strukturen hinweist.
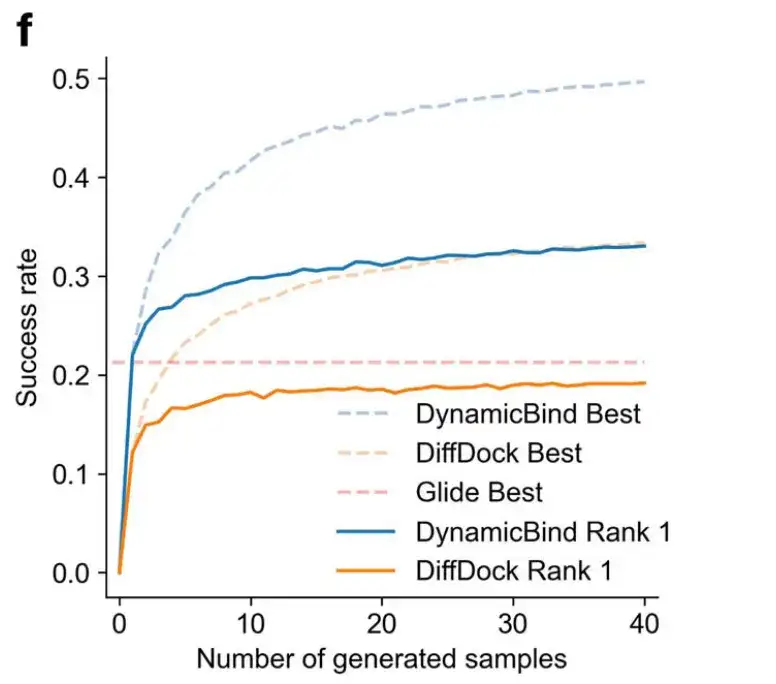
Wie in Abbildung f unten gezeigt, steigt die Erfolgsrate des DynamicBind-Modells bei der Vorhersage der Ligandenposen mit der Anzahl der generierten Proben.
DynamicBind kann ligandenspezifische Proteinkonformationsänderungen erfassen
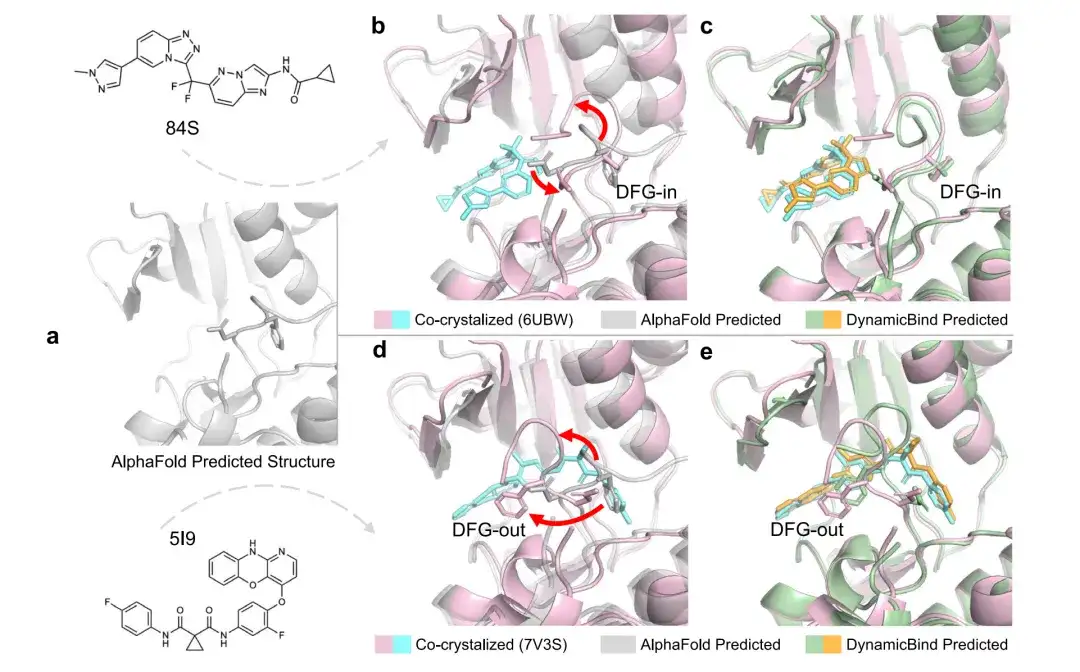
Bei herkömmlichen Docking-Protokollen wird die Proteinkonformationsprobe normalerweise in einem vom Docking-Prozess getrennten Schritt durchgeführt. In vielen Fällen können jedoch zwei verschiedene Liganden in sich gegenseitig ausschließende Proteinkonformationen passen. In früheren Docking-Modellen musste das Protein in die richtige Konformation voreingestellt werden, bevor die geeignete Bindungsposition des Liganden identifiziert werden konnte.Im Gegensatz dazu verwendet DynamicBind die von AlphaFold vorhergesagte Proteinkonformation, um die Proteinkonformation dynamisch anzupassen und die optimale Konformation zu finden, die am besten zum betreffenden Liganden passt, siehe Abbildung a unten.
Die Abbildungen b bis e zeigen den RMSD der von DynamicBind und AlphaFold vorhergesagten Liganden und Taschen in den Strukturen PDB 6UBW und PDB 7V3S. Für PDB 6UBW sagt DynamicBind einen Liganden-RMSD von 0,49 Å und einen Taschen-RMSD von 1,97 Å voraus, während der Taschen-RMSD der AlphaFold-Struktur 9,44 Å beträgt. Für PDB 7V3S sagt DynamicBind einen Liganden-RMSD von 0,51 Å und einen Taschen-RMSD von 1,19 Å (AlphaFold 6,02 Å) voraus.
Die Abbildungen f und g zeigen, wie sich mit UniProt ID markierte Proteine, ausgehend von der gleichen Ausgangsstruktur, nach der Bindung an Typ-I-Inhibitoren allmählich in Richtung der DFG-in-Konformation bewegen und bei der Interaktion mit Typ-II-Inhibitoren zur DFG-out-Konformation tendieren.
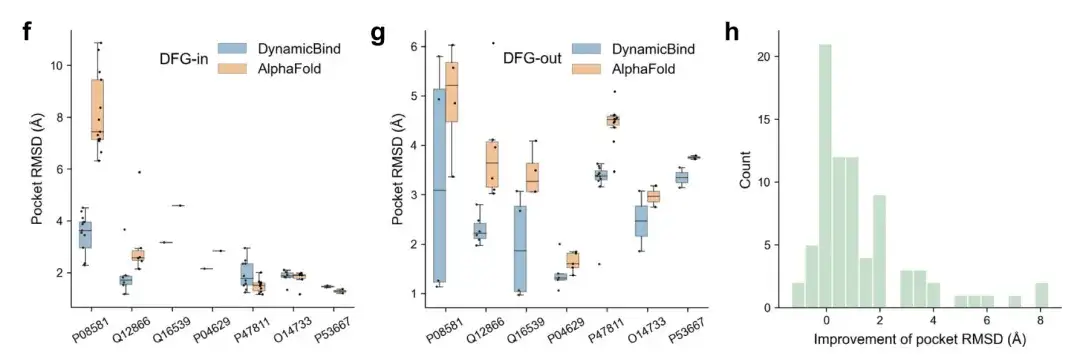
Abbildung h zeigt, dass die meisten der von DynamicBind vorhergesagten Proteinstrukturen einen niedrigeren Taschen-RMSD aufweisen als die ursprüngliche AlphaFold-Struktur.
Die obigen Ergebnisse zeigen, dass DynamicBind in der Lage ist, ligandenspezifische Konformationsänderungen zu erfassen.Das heißt, DynamicBind kann Verbindungen identifizieren, die gut an andere mögliche Konformationen des Proteins binden, selbst wenn die jeweilige Konformation von der ursprünglich bereitgestellten Proteinstruktur abweicht.
DynamicBind deckt mehrere Skalen von Proteinkonformationsänderungen ab
Die Forscher bewerteten DynamicBind anhand von sechs verschiedenen Arten von maßstabsübergreifenden Konformationsänderungen im Bereich von Pikosekunden bis Millisekunden.Wie in der folgenden Abbildung gezeigt, stellt Pink die Kristallstruktur dar, Weiß die AlphaFold-Struktur, Grün die von DynamicBind vorhergesagte Struktur, Cyan den nativen Liganden und Orange den von DynamicBind vorhergesagten Liganden.
Basierend auf dem Vergleich mit der Kristallstruktur,Δpocket RMSD misst den Pocket-RMSD-Unterschied zwischen der vom Modell vorhergesagten Proteinstruktur und der AlphaFold-Struktur.Ein negativer Δpocket RMSD zeigt an, dass die von DynamicBind vorhergesagte Struktur näher an der von AlphaFold vorhergesagten Struktur liegt als an der Kristallstruktur.
Δclash misst den Unterschied in den Kollisionswerten zwischen dem vorhergesagten Protein-Liganden-Paar und dem gepfropften Liganden in der AlphaFold-Struktur.Ein negativer Δclash zeigt weniger Konflikte im vorhergesagten Komplex an.
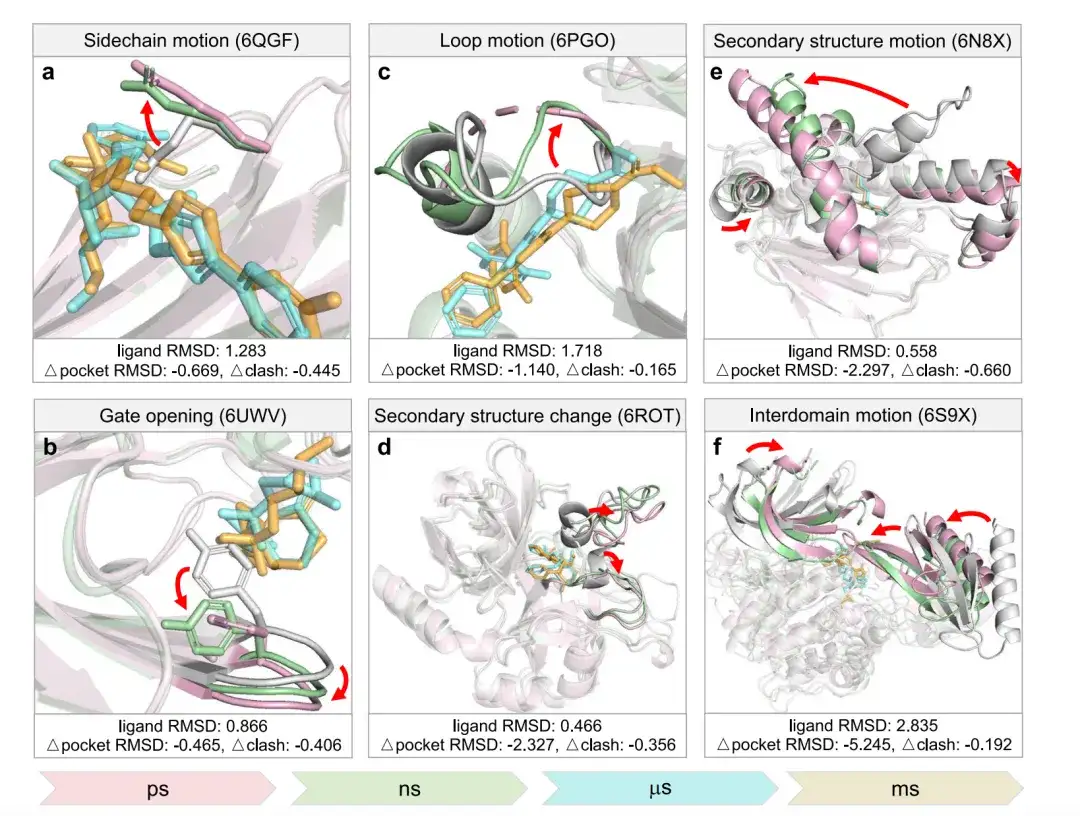
In Abbildung a kollidiert der native Ligand mit einer Seitenkette der darübergelegten AlphaFold-Struktur. Bei der DynamicBind-Vorhersage bewegt sich diese Seitenkette in Richtung der nativen Konformation und löst so den Konflikt. In Abbildung b blockiert ein Tyrosin in der AlphaFold-Struktur einen Teil der Tasche; In der von DynamicBind vorhergesagten Struktur und der nativen Struktur wird dieser Teil der Tasche zugänglich. In Feld c schneidet eine flexible Schleife den Liganden, während sie in der DynamicBind-Vorhersage wegbewegt wird, was mit der nativen Struktur übereinstimmt.
In Abbildung d wird die α-Helix in der Nähe der Ligandenbindungsstelle zu einer Schleife. In Abbildung e erfährt die Sekundärstruktur des Hitzeschockproteins Hsp90α eine große Bewegung, wenn sie von einem geschlossenen in einen offenen Zustand übergeht. In Bild f verdichten sich die beiden AKT1-Kinasedomänen und bilden eine Tasche, die vorher nicht existierte.
Zusammenfassend lässt sich sagen, dass das DynamicBind-Modell verschiedene mit der Ligandenbindung verbundene Konformationsänderungen vorhersagen kann, wenn die Ligandenbindungstasche nicht groß genug ist oder nicht die von AlphaFold vorhergesagte Konformation bildet.
DynamicBind identifiziert kryptische Bindungsstellen
Proteine bilden während dynamischer Prozesse häufig kryptische Taschen, die Stellen freilegen können, an denen Medikamente ansetzen können, die in statischen Strukturen nicht zu finden sind. Dadurch werden Proteine, die zuvor nicht für Medikamente geeignet waren, zu potenziellen Zielen für Medikamente.Anhand einer Fallstudie des SET-Domänen-haltigen Proteins 2 (SEtD2) demonstrierten die Forscher die Nützlichkeit von DynamicBind bei der Aufdeckung dieser kryptischen Taschen.
SETD2, eine Histonmethyltransferase und ein Schlüsselmedikament zur Behandlung des multiplen Myeloms (MM) und des diffusen großzelligen B-Zell-Lymphoms (DLBCL), verfügt über eine kryptische Tasche und ist das Ziel einer hochselektiven Verbindung, EZM0414, die sich derzeit in klinischen Studien der Phase I befindet.
Wie in den Abbildungen a und b unten gezeigt, kokristallisierten alle SETD2-Homologe im Trainingssatz (definiert durch eine Protein-Smith-Waterman-Ähnlichkeit von mehr als 0,4) mit S-Adenosylmethionin (SAM) oder Sinefungin-Analoga, dargestellt durch Linien. Der cyanfarbene Balken zeigt den Liganden EZM0414 von PDB 7TY2 an und der rosa Balken zeigt das Protein an.
In Abbildung c stellt Weiß die AlphaFold-Struktur und ihre Oberfläche dar, wo die kryptischen Stellen blockiert sind, was zu einer großen Anzahl von Konflikten mit dem transplantierten EZM0414 führt.
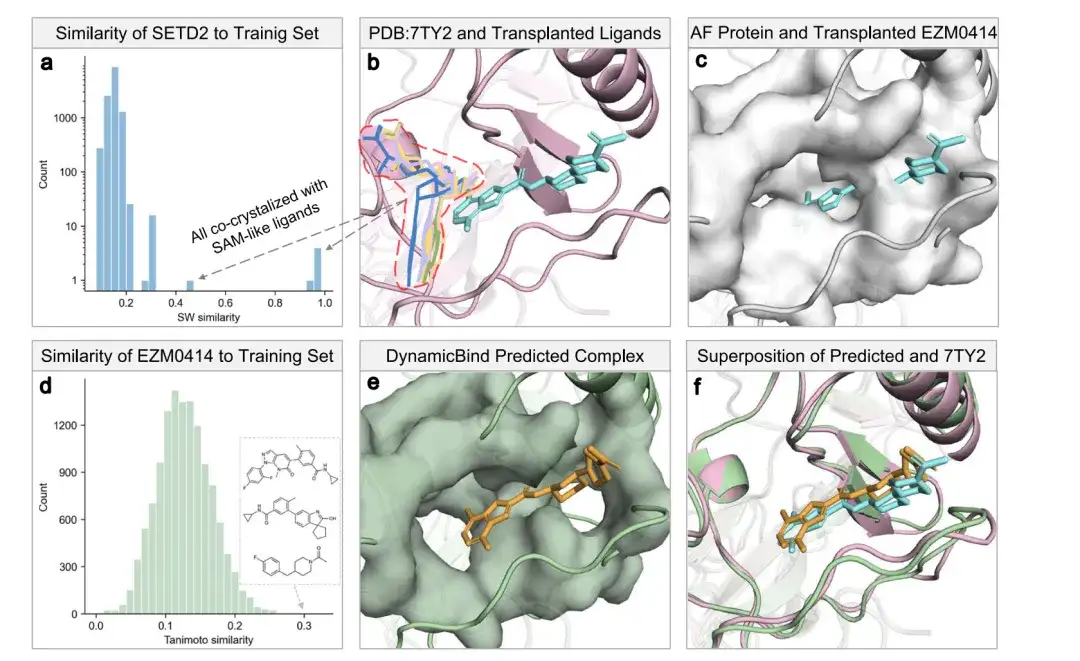
Panel d bestätigt, dass EZM0414 ein unsichtbarer Ligand ist und dass selbst der ähnlichste Tanimoto-Ligand erheblich von EZM0414 abweicht. Abbildung e zeigt die vom DynamicBind-Modell vorhergesagte Protein-Liganden-Komplexstruktur, die die von AlphaFold vorhergesagte SETD2-Struktur und die SMILES-Darstellung von EZM0414 als Eingabe verwendet. Abbildung f zeigt die Überlappung zwischen der von DynamicBind vorhergesagten Protein-Liganden-Komplexstruktur und der Kristallstruktur des SETD2-EZM0414-Komplexes (PDB 7TY2).
Den Ergebnissen zufolge gelang es DynamicBind, die verborgene Tasche dynamisch anzudocken. Dabei wurde nicht nur der Ligand erfolgreich platziert, sondern auch eine geeignetere Taschenkonformation gefunden (der RMSD des erhaltenen Liganden betrug 1,4 Å und der RMSD der Tasche 2,16 Å).
DynamicBind erzielt bessere Ergebnisse beim Arzneimittelscreening bei Antibiotika-Benchmarks
Im zielgerichteten Arzneimittelentdeckungsprozess sind sowohl das Screening potenzieller Arzneimittelkandidaten als auch das Gegenscreening (Identifizierung von Proteinzielen für bestimmte Verbindungen) von entscheidender Bedeutung.Um die Screening-Leistung des DynamicBind-Modells in der Praxis zu bewerten, fügten die Forscher dem Modell ein Modul zur Affinitätsvorhersage hinzu, trainierten es mithilfe experimentell gemessener Bindungsaffinitätsdaten aus dem PDBbind-Datensatz und evaluierten es anhand von Arzneimittel-Screening-Testdaten aus dem 2023 veröffentlichten Antibiotika-Proteom (darunter 12 Proteinziele und fast 3.000 gemessene Aktivitätsdaten).
Wie in Abbildung a unten gezeigt, übertrifft DynamicBind gängige Docking-Methoden wie VINA und DOCK6.9 sowie die beste Rescoring-Methode auf Basis von maschinellem Lernen mit einer durchschnittlichen Fläche unter der Receiver-Operating-Characteristic-Kurve (auROC) von 0,68. Diese Leistungsverbesserung ist auf die dynamische Docking-Funktion von DynamicBind zurückzuführen, die die AlphaFold-Struktur auf einen näher an ihrem nativen Zustand liegenden Zustand verfeinern und so eine genauere Schätzung der Bindungsaffinität erreichen kann.
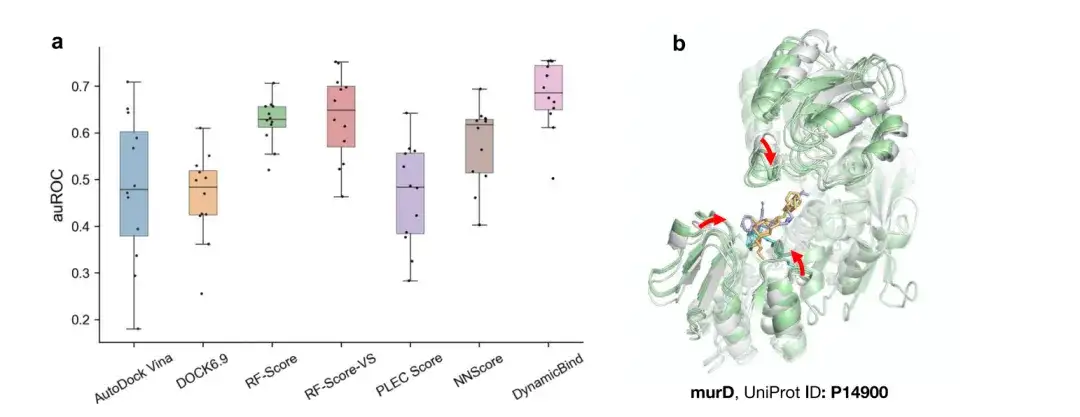
Abbildung b oben zeigt, dass die von DynamicBind vorhergesagte Protein-murD-Struktur den Liganden enger umgibt und mehr Wechselwirkungen bildet, die in der ursprünglichen AlphaFold-Struktur nicht möglich waren.
Diese Ergebnisse zeigen, dass DynamicBind herkömmliche Docking-Methoden und auf Deep Learning basierende starre Docking-Methoden durchweg übertrifft und dass das Modell aufgrund seiner Fähigkeiten zur Vorhersage der Bindungsaffinität großes Potenzial für virtuelle Screening-Anwendungen auf Proteomebene aufweist.
Die Entschlüsselung der komplexen Struktur und Funktion von Proteinen soll zur intelligenten Arzneimittelentdeckung beitragen
Basierend auf der statischen Strukturvorhersage von AlphaFold führt das DynamicBind-Modell auf innovative Weise generative künstliche Intelligenztechnologie ein und löst erfolgreich die Herausforderung der dynamischen Vorhersage komplexer Strukturen. Die Vorhersage dynamischer Veränderungen der Proteinstruktur ist für das Verständnis von Lebensprozessen und die Entwicklung neuer Medikamente von großer Bedeutung. Insbesondere bei der KI-basierten Arzneimittelentwicklung kann es die Genauigkeit und klinische Wirksamkeit des KI-basierten Arzneimittelscreenings erheblich verbessern.
Als einer der Hauptbeteiligten an diesem Forschungsergebnis engagiert sich die Forschungsgruppe von Zheng Shuangjia seit langem intensiv in der übergreifenden Spitzenforschung zu generativer künstlicher Intelligenz und Arzneimittelentwicklung und hat dabei fruchtbare Ergebnisse erzielt.
Am 21. Juni 2024 schlug die Forschungsgruppe von Zheng Shuangjia eine kreuzmodale Lernmethode vor, die gestörte, hochkonzentrierte Zellmikroskopiebilder auf phänotypischer Ebene verwendet, um das Lernen molekularer Repräsentationen zu unterstützen.Mit diesem Ansatz lässt sich effektiv eine Brücke zwischen Molekülen und Charakterisierung schlagen, was für die Arzneimittelentwicklung von großer Bedeutung ist. Die zugehörige Forschung wurde in Advanced Science unter dem Titel „Cross-Modal Graph Contrastive Learning with Cellular Images“ veröffentlicht.
Papieradresse:
https://onlinelibrary.wiley.com/doi/10.1002/advs.202404845
Am 25. Mai 2024 schlug die Forschungsgruppe von Zheng Shuangjia das Multiskalen-Lernframework MUSE vor, das Multiskaleninformationen zwischen Atomstruktur und molekularen Netzwerkskalen effektiv integriert.Demonstriert das Potenzial, die computergestützte Arzneimittelforschung auf andere Maßstäbe auszuweiten. Die entsprechende Forschung wurde in Nature Communications unter dem Titel „A variational expectation-maximization framework for balanced multi-scale learning of protein and drug interactions“ veröffentlicht.
Papieradresse:
https://www.nature.com/articles/s41467-024-48801-4
Am 15. September 2022 entwickelte die Forschungsgruppe von Zheng Shuangjia einen generativen intelligenten Arzneimitteldesign-Algorithmus für schwer medikamentenfähige Zielmoleküle und entwarf in kurzer Zeit PROTAC-Leitverbindungen.Dies wurde durch Tierversuche bestätigt und zeigt das enorme Potenzial der Integration von Informationstechnologie und Biotechnologie. Diese Reihe von Ergebnissen hat positive Zitate und Bewertungen von führenden Forschungsgruppen auf diesem Gebiet erhalten, darunter das Google DeepMind AlphaFold-Team und das Team von Professor David Baker an der University of Washington. Die entsprechende Forschung wurde in Nature Machine Intelligence unter dem Titel „Accelerated rational PROTAC design via deep learning and molecular simulations“ veröffentlicht.
Papieradresse:
https://www.nature.com/articles/s42256-022-00527-y
Am 14. Februar 2020 schlug die Forschungsgruppe von Zheng Shuangjia ein quasi-visuelles Frage-Antwort-System vor, das auf einem End-to-End-Deep-Learning-Framework basiert.Durch die Identifizierung der Wechselwirkung zwischen Arzneimitteln und Proteinen wird die Arzneimittelentdeckung effektiv erleichtert. Die entsprechende Forschung wurde in Nature Machine Intelligence unter dem Titel „Predicting drug–protein interaction using quasi-visual question answering system“ veröffentlicht.
Papieradresse:
https://www.nature.com/articles/s42256-020-0152-y
Basierend auf ihrem Verständnis der übergreifenden Spitzenforschung zu generativer künstlicher Intelligenz und Arzneimittelentwicklung konzentriert sich die Forschungsgruppe von Zheng Shuangjia auf die intelligente Entwicklung von Arzneimitteln für Stoffwechsel- und Alterungskrankheiten, erstellt ein neues Arzneimittelentwicklungsmodell, das IT und BT integriert, und engagiert sich dafür, einen größeren Beitrag zur durchgängigen intelligenten Arzneimittelentdeckung zu leisten.
Quellen:








