Command Palette
Search for a command to run...
Neue Ergebnisse Aus Der Maßgeblichen Fachzeitschrift Cell Discovery! Das Team Um Hong Liang Von Der Shanghai Jiao Tong University Schlug Ein CPDiffusion-Modell Für Die Ultra-kostengünstige Und Vollautomatische Entwicklung Funktioneller Proteine Vor
Proteine sind die Hauptakteure des Lebens und die Beziehung zwischen ihrer Struktur und Funktion ist seit jeher ein zentrales Forschungsthema im Bereich der Biowissenschaften. In den letzten Jahren konnte das Modell mit dem Aufkommen des Deep Learning und seinen leistungsstarken Datenverarbeitungsfunktionen die Zuordnungsbeziehung zwischen Proteinsequenz, -struktur und -funktion erlernen und neue Proteine mit höherer Stabilität, stärkerer Bindungsaffinität und höherer Enzymaktivität entwerfen, wodurch die Effizienz des Proteindesigns erheblich verbessert und die F&E-Kosten effektiv gesenkt werden können.
Allerdings erfordern bestehende Methoden normalerweise das Trainieren eines Modells mit extrem großen Parametern auf einem umfangreichen Datensatz, was sich nur schwer auf bestimmte Proteine mit seltenen homologen Sequenzen verallgemeinern lässt und oft nur Proteine mit relativ einfacher Struktur und Funktion erzeugen kann. Darüber hinaus zeigt die experimentelle Überprüfung, dass die entworfenen Proteine im Allgemeinen eine geringere Aktivität aufweisen und dass solche, die Wildtyp-Proteine übertreffen können, selten sind.
In diesem Zusammenhang haben Zhou Bingxin, ein Assistenzforscher in der Forschungsgruppe von Hong Liang von der School of Natural Sciences/School of Physics and Astronomy/Zhangjiang Institute for Advanced Studies/School of Pharmacy der Shanghai Jiao Tong University, und andere ein Rahmenmodell für Diffusionswahrscheinlichkeit namens CPDiffusion entwickelt.Dieses Framework kombiniert mehrere Generierungsbedingungen wie die Struktur des Proteinrückgrats und aktive Stellen und kann die implizite Abbildungsbeziehung zwischen Protein-Sequenz, Struktur und Funktion mit sehr geringem Trainings- und Datenaufwand erlernen und dann verschiedene Protein-Sequenzen generieren. Diese generierten Sequenzen können den Test bei der Nassexperimentverifizierung mit einer sehr hohen Erfolgsrate bestehen.
Es ist erwähnenswert, dass der Trainings- und Inferenzprozess von CPDiffusion fast keine Expertenanleitung erfordert.Es kann hochkonservierte Regionen automatisch identifizieren und dann basierend auf den Funktionen der konservierten Regionen weitere Änderungen in nicht konservierten Regionen einführen, um die Vielfalt der generierten Sequenzen zu erhöhen. Die Studie mit dem Titel „Ein bedingtes Proteindiffusionsmodell erzeugt künstliche programmierbare Endonukleasesequenzen mit erhöhter Aktivität“ wurde in Nature’s Cell Discovery veröffentlicht.
Forschungshighlights:
* Im Rahmen der Studie wurden die Endonukleasen KmAgo und PfAgo erfolgreich entwickelt und erzeugt, deren DNA-Spaltungsaktivität um mehr als das Zehnfache erhöht wurde, deutlich höher als die Aktivität der derzeit entdeckten Wildtyp-Proteine bei mittleren Temperaturen.
* Diese Studie kann Hunderte von Aminosäuren gleichzeitig verändern und bietet so mehr Möglichkeiten für die Protein-Engineering-Forschung
* Die vielfältige Generierung neuer Proteinsequenzen kann auch die Datenbank der Proteinfamilien erweitern und Wissenschaftlern umfangreichere Forschungsressourcen bieten

Link zum Artikel:
https://www.nature.com/articles/s41421-024-00728-2
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Stellen Sie die Stichprobenvielfalt sicher und vermeiden Sie Datenverzerrungen
Um die Zuordnungsbeziehung zwischen Proteinsequenz-Struktur-Funktion zu lernen,Das CPDiffusion-Modell wurde mit 20.000 Wildtyp-Proteinen von CATH 4.2 trainiert. Darüber hinaus fügten die Forscher dem Trainingssatz 694 pAgos-Proteine hinzu, um das Verständnis des Modells für die Merkmale der zu generierenden Proteine zu verbessern.
Diese Proteine stammen aus der pAgo-Proteinfamilie, die in früheren Studien zusammengestellt wurde, darunter kurze, lange A- und lange B-pAgo-Proteine, wodurch die Vielfalt der ausgewählten Proben sichergestellt wird, um mögliche Probleme mit Datenverzerrungen zu reduzieren. Darüber hinaus sind die meisten WT-Proteine im Datensatz mesophile pAgos und nur wenige Long-A-pAgo-Proteine sind thermophil.
Modellarchitektur: 6-stufiges automatisiertes pAgo-Proteindesign
Um die Wirkung der CPDiffusion auf die Erzeugung funktioneller Proteine zu überprüfen, konzentrierten sich die Forscher auf das pAgo-Protein. Das pAgo-Protein ist eine Endonuklease, die eine wichtige Rolle im DNA-Interferenzprozess von Prokaryoten spielt. Es kann bestimmte einzelsträngige DNA- oder RNA-Sequenzen spezifisch erkennen und schneiden und hat einen breiten Anwendungswert im Bereich der Diagnostik. Darüber hinaus haben pAgo-Proteine eine hohe Affinität zu Substraten und können Zielsequenzen spezifisch erkennen, was sie zu wichtigen Werkzeugen für die Bildgebung und Genom-Editierung macht.
Forscher nutzten das CPDiffusion-Framework, um neue pAgo-Proteine zu entwerfen.Wie in Abbildung a unten gezeigt,Zunächst werden die Sequenz und Informationen des Eingabeproteins (Original pAgo) in eine grafische Darstellung umgewandelt, die die molekularbiochemischen und topologischen Eigenschaften des Proteins auf Aminosäureebene anzeigt.Wie in Abbildung b gezeigt,Das Protein tritt in die Vorwärtsdiffusionsphase ein, in der jeder Aminosäuretyp im ursprünglichen Protein einer bestimmten Substitutionswahrscheinlichkeitsmatrix folgt und in einer Reihe von Schritten (T-Schritten) zerstört wird, bis die gesamte Sequenz gleichmäßig verteilt ist.
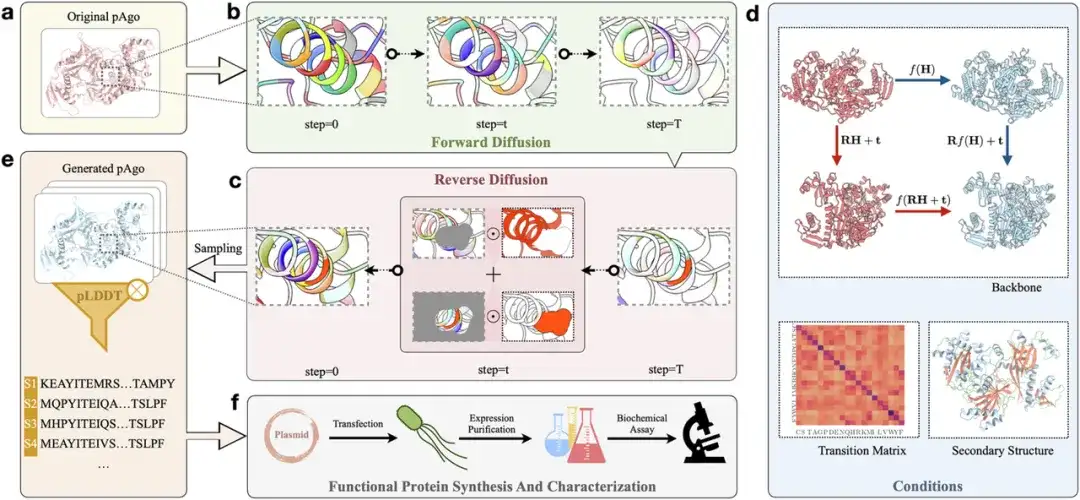
Wie in Abbildung c oben gezeigt,In der Phase der umgekehrten Diffusion wählten die Forscher zufällig Aminosäuren aus 20 gleichmäßig verteilten Aminosäuretypen aus und entfernten dann schrittweise das Rauschen aus der Proteinsequenz.Wie in Abbildung d oben gezeigt,Während des Rauschunterdrückungsprozesses verwenden Forscher bestimmte Bedingungen (wie etwa die Wildtyp-Rückgratstruktur des Zielproteins, die Sekundärstruktur und die Aminosäuresubstitutionsmatrix basierend auf dem Wildtyp-Protein), um den Prozess zu steuern. Um sicherzustellen, dass das Modell die in der dreidimensionalen Struktur des Proteins implizite Äquivarianz erlernen kann, verwendeten die Forscher eine äquivariante Graph-Faltungsschicht, um die Ausbreitungsfunktion anzupassen. Das Modell generiert dann eine gemeinsame Wahrscheinlichkeitsverteilung für jede Aminosäureposition auf dem Proteinrückgrat. Durch die Stichprobennahme der erlernten Verteilung können Forscher die entsprechende Proteinsequenz erhalten (generiertes pAgo).Wie in Abbildung e oben gezeigt.
Anschließend verwendeten die Forscher AlphaFold2, um eine Strukturvorhersage für die generierten Sequenzen durchzuführen, und filterten geeignete Sequenzen heraus, indem sie Indikatoren wie RMSD und pLDDT auswerteten. endlich,Wie in Abbildung f unten gezeigt,Diese geeigneten Sequenzen werden im Labor Nassexperimenten (Synthese, Charakterisierung und Evaluierung) unterzogen, um ihre tatsächlichen Eigenschaften wie Expressionsniveau, Enzymaktivität und thermische Stabilität weiter zu bestätigen.
Experimentelles Fazit: Das neue Protein hat eine stärkere Aktivität und thermische Stabilität
Die Forscher verwendeten mesophile pAgo-Proteine (wie KmAgo) und thermophile pAgo-Proteine (wie PfAgo) als Kandidatenproteine und generierten darüber hinaus zwei Gruppen neuer Proteinsequenzen. Wie in der folgenden Abbildung gezeigt, konnten die Forscher mithilfe des Generierungs- und Screening-Frameworks CPDiffusion erfolgreich 27 neue künstliche KmAgos (Km-APs) und 15 neue künstliche PfAgos (Pf-APs) generieren. Diese neu generierten Proteine haben eine Sequenzidentität von 50%-70% im Vergleich zur ursprünglichen Wildtyp-Vorlage (WT) und eine Sequenzidentität von weniger als 40% im Vergleich zu anderen Nicht-Vorlagen-WT-Proteinen (d. h. anderen WT-Proteinen in der NCBI-Datenbank).
* KmAgo ist ein mesophiles Enzym mit relativ geringer DNA-Spaltungsaktivität im Wildtyp, was sein Potenzial für praktische Anwendungen begrenzt
* PfAgo ist ein ultra-thermogenes Enzym. Der Wildtyp weist eine höhere DNA-Spaltungsaktivität auf, funktioniert aber normalerweise nur bei hohen Temperaturen. Mit sinkenden Temperaturen nimmt auch die Aktivität ab.
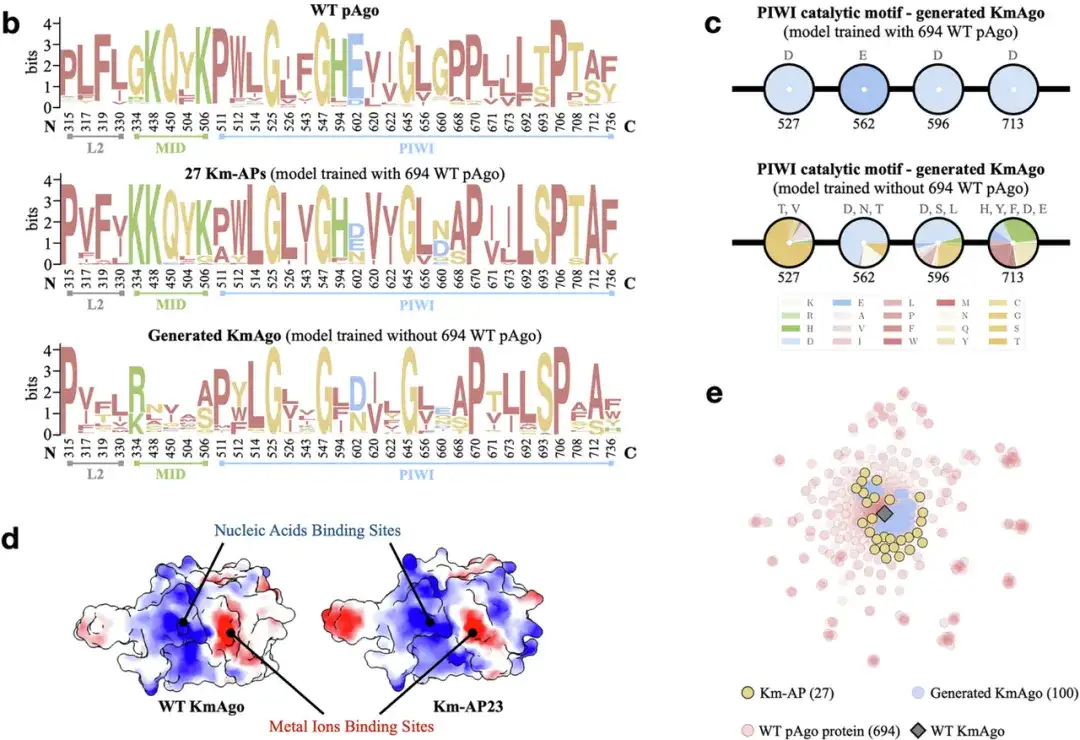
Es ist erwähnenswert, dassDer Trainings- und Inferenzprozess von CPDiffusion erfordert fast keine Expertenanleitung.Es kann hochkonservierte Regionen automatisch identifizieren und dadurch basierend auf den Funktionen der konservierten Regionen mehr Änderungen in nicht konservierten Regionen einführen, um die Vielfalt der generierten Sequenzen zu erhöhen.
Durch verschiedene experimentelle Überprüfungen, wie in der Abbildung unten dargestellt, fanden die Forscher heraus, dass in den neuen Sequenzen, die für KmAgo generiert wurden,Alle Sequenzen wurden ausgedrückt. Fast 901 TP3T-Sequenzen wiesen eine DNA-Spaltungsaktivität auf und mehr als 701 TP3T-Sequenzen zeigten eine höhere Aktivität als der Wildtyp. Unter ihnen hatte der leistungsstärkste neue KmAgo eine fast neunmal höhere Aktivität als der Wildtyp KmAgo. Darüber hinaus war die thermische Stabilität einiger Km-APs im Vergleich zum Wildtyp-KmAgo verbessert.
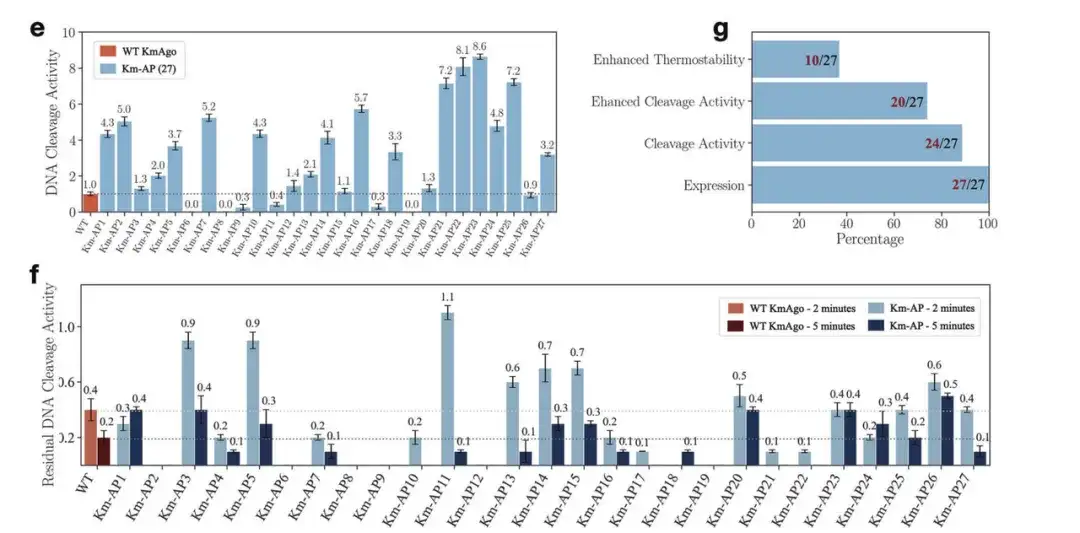
e: DNA-Spaltungsaktivität von 27 Km-APs bei 37 °C
g: Die Anzahl der Proteine, die in 27 Km-APs unterschiedliche Rollen spielen
f: DNA-Spaltungsaktivitäten von WT KmAgo und 27 Km-APs nach Inkubation bei 42 °C für 2 und 5 Minuten.
Wie in der Abbildung unten gezeigt,Von den 15 neuen Sequenzen, die für PfAgo generiert wurden, wurden alle exprimiert und zeigten eine einzelsträngige DNA-Spaltaktivität. Unter ihnen senkte das leistungsstärkste neue PfAgo nicht nur die Schmelztemperatur des Wildtyp-PfAgo von etwa 100 °C auf etwa 50 °C, sondern seine Aktivität zur Spaltung einzelsträngiger DNA war bei 45 °C auch doppelt so hoch wie die des Wildtyp-PfAgo bei 95 °C und 11-mal so hoch wie die des Wildtyp-KmAgo bei mittlerer Temperatur.
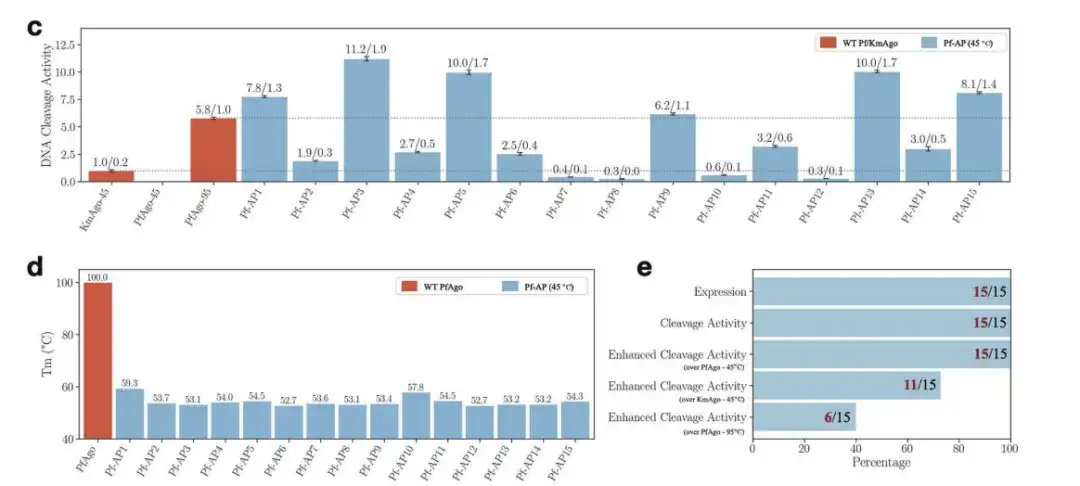
c: DNA-Spaltungsaktivität von 15 Pf-APs bei 45 °C
d: Schmelztemperatur von WT PfAgo und Pf-AP
e: Die Anzahl der Sequenzen, die bei den 15 Pf-APs unterschiedliche Rollen spielen
Zusammenfassend kann CPDiffusion als leistungsstarkes neues Protein-Sequenz-Design-Tool verwendet werden, das automatisch von Wildtyp-Funktionsproteinen lernen und leistungsfähigere komplexe Protein-Sequenzen entwerfen kann, wodurch die vorhandene Proteindatenbank bereichert wird und mehr Möglichkeiten für das Protein-Engineering-Design geschaffen werden.
KI verändert die Zukunft des Protein-Engineerings
Der Einsatz von KI zur Entschlüsselung der Geheimnisse von Proteinen ist für die Digitalisierung der Life-Science-Forschung von zentraler Bedeutung. In diesem Wettlauf um die Erforschung der Essenz des Lebens machen chinesische Forschungsteams kontinuierliche Fortschritte und leisten ihren Beitrag. Als einer der herausragenden Vertreter auf diesem GebietProfessor Hong Liang, der korrespondierende Autor dieser Studie, und sein Forschungsteam konzentrieren sich seit langem auf die durch KI gesteuerte Proteinmodifikation und die unterstützte Arzneimittelentwicklung.Zu den spezifischen Forschungsinhalten zählen unter anderem die Vorhersage und Optimierung von Proteinstrukturen, die gezielte Modifikation und Gestaltung von Proteinen, die Entwicklung und Optimierung von Hilfsmedikamenten usw. Das Team hat fruchtbare Ergebnisse erzielt. Bisher wurden insgesamt 77 Artikel veröffentlicht, viele davon schafften es an die Spitze der Fachzeitschrift „Nature“.
Homepage der Forschungsgruppe von Professor Hong Liang:
https://ins.sjtu.edu.cn/people/lhong/index.html
Seit 2021 versucht das Team von Professor Hong Liang, KI beispielsweise auf den Proteinbereich anzuwenden.Erstellen Sie proprietäre Modelle im Bereich des Protein-Engineerings, um Sequenzen für die Funktion von Anfang bis Ende zu entwerfen.Sie haben mit dem Forscher Tan Pan vom Shanghai Artificial Intelligence Laboratory zusammengearbeitet, um eine Feinabstimmungstrainingsmethode FSFP basierend auf dem Protein-Vortrainingsmodell vorzuschlagen. Mit dieser Methode kann das Protein-Vortrainingsmodell mithilfe von nur 20 zufälligen Nass-Experimentdaten effizient trainiert werden, wodurch die Positivitätsrate der Vorhersage einzelner Punktmutationen des Modells erheblich verbessert wird. Es kann zum Lernen der Proteinanpassungsfähigkeit anhand kleiner Stichproben verwendet werden und hat in praktischen Anwendungen großes Potenzial gezeigt.
Das Team von Professor Hong Liang entwickelte außerdem ein mikroumgebungsbewusstes Graph-Neuralnetzwerk namens ProtLGN.Es kann vorteilhafte Aminosäuremutationsstellen aus der dreidimensionalen Struktur von Proteinen lernen und vorhersagen und die Gestaltung von Einzelstellenmutationen und Mehrfachmutationen mit unterschiedlichen Funktionen leiten. Die experimentellen Ergebnisse zeigten, dass mehr als 401 TP3T ProtLGN-entwickelte Einzelpunktmutantenproteine ihre Wildtyp-Gegenstücke übertrafen.
Weitere Details: Ohne experimentelle Daten zur Steuerung der gerichteten Proteinevolution veröffentlichte die Forschungsgruppe von Hong Liang von der Shanghai Jiaotong University das mikroumgebungsbewusste Graph-Neuralnetzwerk ProtLGN
Darüber hinaus führten sie einen einfachen, effizienten und skalierbaren Adapter ein, den SES-Adapter,Durch die Kombination von Einbettungen von Proteinsprachenmodellen mit Einbettungen von Struktursequenzen zur Erstellung strukturbewusster Darstellungen kann die Leistung von Proteinsprachenmodellen erheblich verbessert werden.
Die oben genannte Forschung zeigt das enorme Potenzial von Deep Learning im Proteindesign. Es besteht kein Zweifel, dass die Protein-Engineering-Forschung mit der weiteren Anwendung der Deep-Learning-Technologie im Proteinbereich einen breiteren Entwicklungsraum eröffnen wird.
Quellen:
https://mp.weixin.qq.com/s/a4gsV4yjzKnW4u6Vtl8LiQ
https://ins.sjtu.edu.cn/article








