Command Palette
Search for a command to run...
Vollständige Demontage Von AlphaFold 3, Zhong Bozitao Von Der Shanghai Jiaotong University: Extreme Datennutzung Zur Vorhersage Aller Biomolekularen Strukturen Mit Atomarer Präzision, Aber Nicht Perfekt

AlphaFold 3, das die Strukturen und Wechselwirkungen aller Biomoleküle mit „atomarer Präzision“ vorhersagen kann, hat unmittelbar nach seiner Veröffentlichung breite Diskussionen in der Branche ausgelöst. Am 13. August fand an der Sommerschule für KI für Bioengineering der Shanghai Jiao Tong University statt.Dr. Zhong Bozitao verarbeitete systematisch seine Lernerfahrungen zum Thema „AlphaFold 3: Prinzipien, Anwendungen und Perspektiven“ und analysierte ausführlich viele relevante Forschungsergebnisse aus der wissenschaftlichen Forschungsgemeinschaft, um seine tiefen Einblicke in AlphaFold 3 mit allen zu teilen.HyperAI hat den Kerninhalt der Rede organisiert, ohne die ursprüngliche Absicht zu verletzen. Nachfolgend finden Sie die Abschrift der Rede.

Mit dem Schwerpunkt auf der Vorhersage von Proteinstrukturen sprechen wir heute über AlphaFold 3, das derzeit führende Tool zur Vorhersage von Protein- und biologischen Molekularstrukturen und noch umfangreicher.Der Status von AlphaFold 3 ist offensichtlich.
Die Proteinsynthese beginnt mit der DNA-Transkription, die dann die genetische Information auf RNA überträgt, die dann in Protein übersetzt und weiter in sekundäre, tertiäre und quaternäre Strukturen gefaltet wird. Die meisten Proteine falten sich in einzigartige Konformationen und die für die Struktur erforderlichen Informationen sind in der Aminosäuresequenz kodiert. Daher sagen wir oft: Die Sequenz bestimmt die Struktur und die Struktur bestimmt die Funktion.Die Vorhersage der Proteinstruktur ist für das Verständnis biologischer Funktionen von entscheidender Bedeutung.
Durchbruch bei AlphaFold 3: Innovative Modellarchitektur und verbesserte Datennutzung
Vergleich der Modellarchitekturen AlphaFold 3 und AlphaFold 2
In der Vergangenheit hat AlphaFold 2 andere Algorithmen bei der Vorhersage von Proteinstrukturen direkt „geschlagen“.Seine Kernarchitektur lässt sich in drei Hauptteile zusammenfassen, wie in der folgenden Abbildung dargestellt: Der erste Teil, das MSA & Template-Modul im blauen Kasten, ist für das Sammeln und Integrieren von Informationen zur Mehrfachsequenzalignmentierung (MSA) und Template-Struktur als Eingabedaten für das Modell verantwortlich. Der zweite Teil, das Evoformer-Modul im grünen Kasten, ist dafür verantwortlich, die koevolutionären Informationen in Multisequenzorganisationen zu verstehen, die gesammelten Informationen zu verfeinern und zu verarbeiten und sie an das Strukturmodul im violetten Kasten im dritten Teil weiterzuleiten.
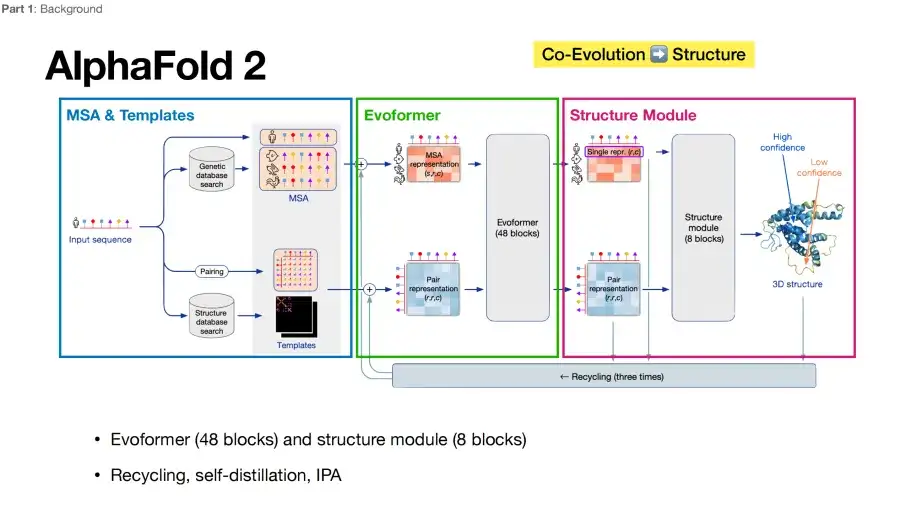
Aus der Perspektive des Deep Learning spielt Evoformer die Rolle eines Encoders, während das Structure Module einem Decoder entspricht.AlphaFold 2 hat vor allem aufgrund seiner End-to-End-Optimierungsfunktionen, die eine direkte Abbildung von der Sequenzeingabe zur Strukturausgabe ermöglichen, viel Anerkennung erhalten.
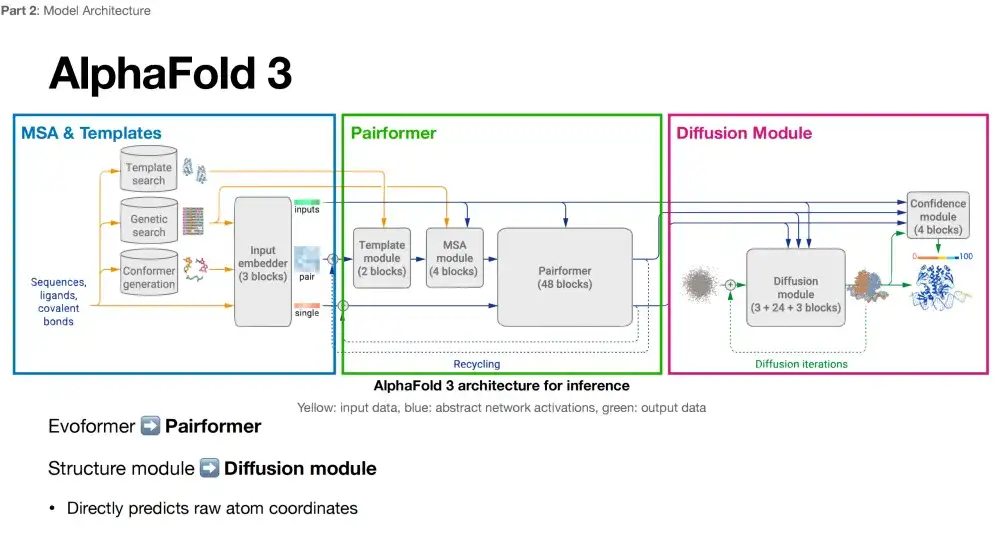
Es wird allgemein angenommen, dass die Änderungen in der Modellarchitektur von AlphaFold 3 nicht so groß sind wie gedacht. Auch sein Modellrahmen besteht aus drei Hauptteilen. Der Vergleich zwischen den einzelnen Teilen und AlphaFold 2 ist wie folgt:
Teil 1: Die Ähnlichkeit hochhalten
Wie in der folgenden Abbildung gezeigt, ist durch Vergleich der Architekturdiagramme von AlphaFold 3 und AlphaFold 2 ersichtlich, dass der erste Teil von AlphaFold 3 (im blauen Feld) noch MSA und Template enthält und zusätzlich den Conformer-Generationslink einführt.
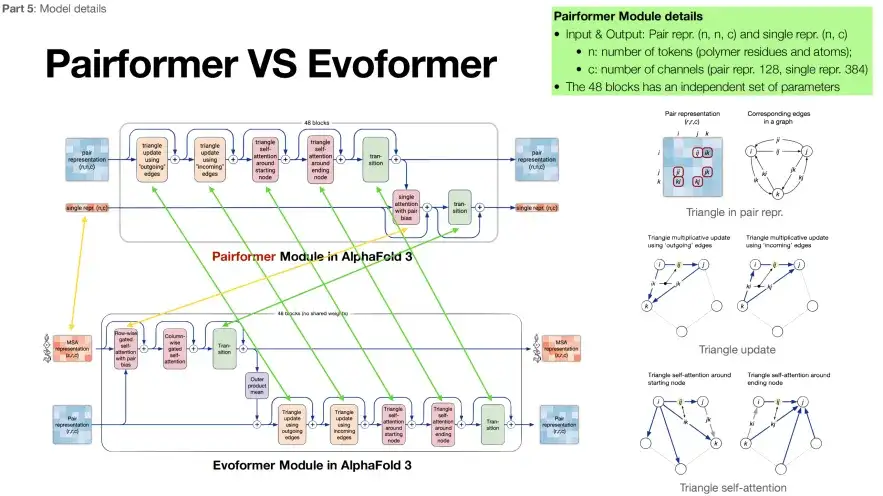
Teil 2: Verringerung der Abhängigkeit von MSA-Sequenzen
Der zweite Teil von AlphaFold 3 (im grünen Kasten) heißt Pairformer. Seine Struktur ist im Wesentlichen Evoformer sehr ähnlich, aber die Anzahl der MSA-Module ist auf 4 reduziert. Wie in der folgenden Abbildung gezeigt, zeigen die grünen Pfeile den gleichen Inhalt in den beiden Modulen an und die gelben Pfeile zeigen Unterschiede an. Wie Sie sehen können,AlphaFold 3 legt mehr Wert auf die Zielproteinsequenz und verlässt sich weniger auf die MSA-Sequenz.
Darüber hinaus glauben wir, dass der Grund für die starke Leistung von AlphaFold 3 bei mehreren Aufgaben darin liegen könnte, dass es seine Abhängigkeit von der multiplen Sequenzalignmentierung (MSA) reduziert.Wie in der folgenden Abbildung dargestellt, zeigt die rechte Seite die Auswirkungen von MSA auf die Leistung von AlphaFold 2: Wenn die Anzahl der MSAs zunimmt, flacht die Verbesserung der Leistung von AlphaFold 2 nach Überschreiten eines bestimmten Schwellenwerts (rosa Linie) tendenziell ab. Wie aus dem mittleren Teil der Abbildung unten ersichtlich ist, ist der Einfluss von MSA auf AlphaFold 3 im Vergleich zu AlphaFold 2 abgeschwächt (die Kurve schwankt sehr wenig).
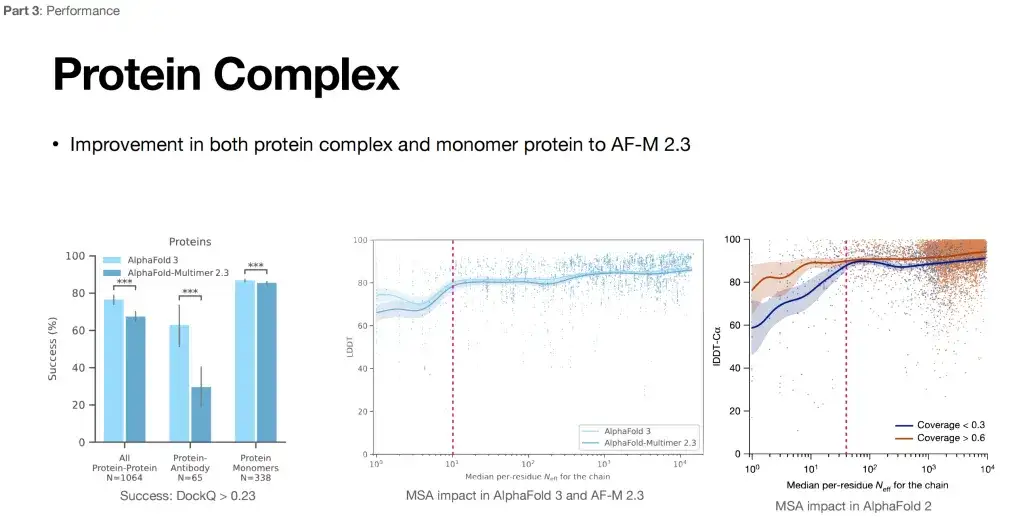
Darüber hinaus erfordert die Reifung von Antikörpern häufig einen Hypermutationsprozess in vivo. MSA-Informationen sind für die Vorhersage ihrer Struktur nur begrenzt hilfreich und es ist schwierig, gepaarte MSA-Informationen für Proteine und ihre Komplexe zu finden. Aus dieser Sicht könnte die Erweiterung des Anwendungsbereichs von AlphaFold 3 die Verringerung seiner Abhängigkeit von MSA sein.
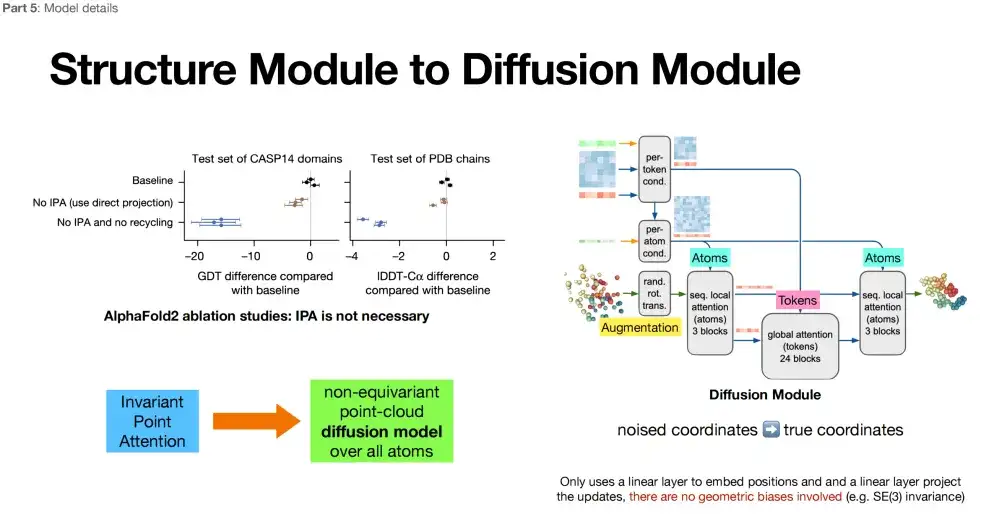
Teil III: Generierung einer Allatomstruktur + Entfernung der Stereorotationsinvarianz
Der dritte Teil von AlphaFold 3 (im violetten Kasten) übernimmt das Diffusionsmodell, das ebenfalls zur Kategorie der Strukturmodule gehört. Der Unterschied besteht darin, dass das Diffusionsmodell die wiederholte iterative Optimierung im Strukturmodul durch einen neuen Mechanismus namens Diffusionsmodell ersetzt.
*Diffusionsmodell: Fügen Sie dem Modell Rauschen hinzu (vorwärts), lassen Sie das Modell Rauschen beseitigen (rückwärts), lernen Sie den umgekehrten Prozess und generieren Sie eine ähnliche Datenverteilung.
Wie in der folgenden Abbildung gezeigt, erreicht AlphaFold 3 im dritten Teil eine Strukturgenerierung auf rein atomarer Ebene. Atome als Grundbausteine von Molekülen können reichhaltigere physikalische Informationen enthalten, was bedeutet, dass AlphaFold 3 bei der Vorhersage von Proteinstrukturen möglicherweise tiefere physikalische Gesetze erfassen kann. Darüber hinaus verzichtet AlphaFold 3 auf die in AlphaFold 2 betonte Stereorotationsinvarianz. Nach dem Löschen der zusätzlichen Architektur dieser Funktion in AlphaFold 2 stellten die Forscher fest, dass das Design des Modells (Diffusionsmodul) freier wurde.
AlphaFold 3 verbessert die Datennutzung
Die Proteindatenressourcen sind begrenzt, aber AlphaFold 3 vergrößert nicht nur den Datensatz, sondern verbessert auch die Datennutzung.Insbesondere nähert sich AlphaFold 3 im Vergleich zum millionenschweren Datensatz von AlphaFold 2 direkt der Milliardenebene und der Trainingssatz ist größer. Darüber hinaus enthält sein Trainingssatz neben den Daten im PDB auch eine große Menge anderer Daten. Beispielsweise sind die von AlphaFold 2 vorhergesagten Strukturdaten als Erweiterung des Trainingssatzes genauer. Der spezifische Trainingssatz ist in der folgenden Abbildung dargestellt:

AlphaFold 3 erreicht einen enormen Sprung im Anwendungsbereich
Die größte Veränderung von AlphaFold 3 besteht darin, dass es einen qualitativen Sprung in seinem Anwendungsbereich erreicht hat.In der Vergangenheit konzentrierte sich AlphaFold 2 eher auf die Vorhersage von Aminosäurestrukturen, während AlphaFold 3 Strukturen auf atomarer Ebene direkt vorhersagen kann. Seine Funktionserweiterung spiegelt sich insbesondere in den folgenden vier Aspekten wider:
* Kann Liganden genau vorhersagen, d. h. die Bindungsstellen kleiner Moleküle in Proteinen vorhersagen;
* Kann die Struktur von Proteinkomplexen vorhersagen;
* Kann die posttranslationalen Modifikationsstrukturen von Proteinen und Nukleinsäuren vorhersagen;
* Kann die Strukturen von DNA und RNA sowie die Strukturen von Komplexen aus DNA/RNA und Proteinen vorhersagen.
AlphaFold 3 verändert das Feld der Liganden-Andockung
Der größte Einfluss von AlphaFold 3 auf das wissenschaftliche Feld liegt unter anderem in der Verbesserung der Liganden-Docking-Aufgabe.Wie in der folgenden Abbildung dargestellt, werden die Erfolgsraten verschiedener Deep-Learning-Algorithmen in 4 verschiedenen Ligand-Docking-Aufgaben im Rahmen des PostBusters-Benchmarks bewertet. Es zeigt sich, dass AlphaFold 3 unter der Voraussetzung unbekannter Taschen und struktureller Vorkenntnisse die höchste Erfolgsrate, d. h. 76,4%, erzielen kann.
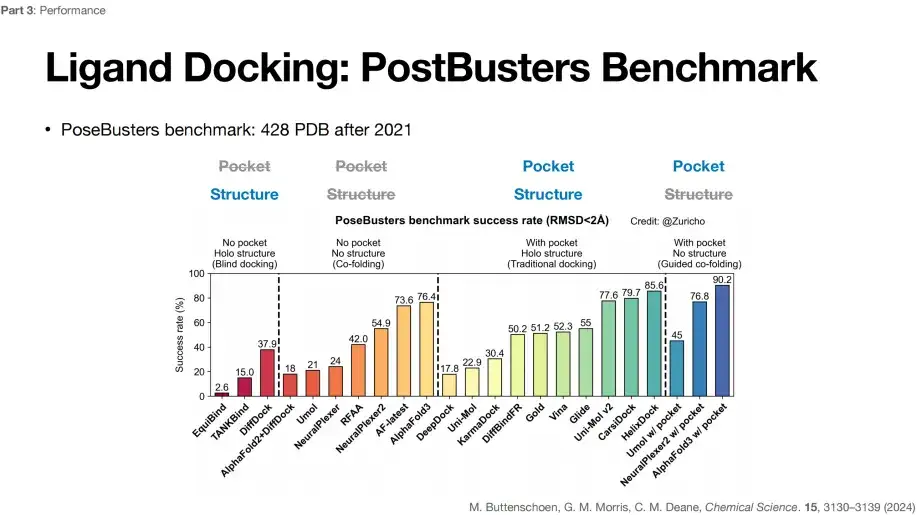
PostBusters Benchmark wählt 428 PDB-Daten aus dem Jahr 2021 und später aus
Der Standard für den Erfolg einer Aufgabe ist, dass die Abweichung zwischen der vorhergesagten Andockposition des kleinen Moleküls und der tatsächlichen Andockposition weniger als 2 Å beträgt
Wie in der obigen Abbildung gezeigt, kann DiffDock beim ersten Typ der Blind-Docking-Aufgabe mit unbekannter Taschenposition und bekannter Proteinstruktur (keine Tasche, Holo-Struktur) die höchste Erfolgsrate von 37,9% erzielen.
Bei der zweiten Art von Co-Faltungsaufgabe (kleine Moleküle und Proteinstrukturen werden gefaltet), mit unbekannten Taschenpositionen und unbekannten Proteinstrukturen (keine Tasche, keine Struktur), sank die Erfolgsrate der kombinierten Vorhersage von AlphaFold 2+DiffDock auf 18%,Darüber hinaus erreichte AlphaFold 3 die höchste Erfolgsrate von 76,4%, was zeigt, dass AlphaFold 3 nicht nur genaue Vorhersagen liefert, sondern auch nicht auf Vorkenntnissen zu Taschen und Strukturen angewiesen ist.
In der dritten Kategorie traditioneller Docking-Aufgaben sind die Taschenposition des kleinen Moleküls und die Proteinstruktur bekannt (Mit-Tasche, Holo-Struktur), d. h. die Tasche liegt frei. Gold erreichte eine Erfolgsrate von 51,2%, Vina zeigte eine Erfolgsrate von 52,3% und Glide stieg auf 55%. Auch andere Deep-Learning-Algorithmen können relativ gute Werte erreichen, was darauf schließen lässt, dass die Erfolgsquote durch die Tasche beeinflusst wird.
In der vierten Kategorie der Aufgaben zum geführten gemeinsamen Falten mit bekannten Taschenpositionen und unbekannten Proteinstrukturen (mit Tasche, ohne Struktur) wurde die Erfolgsrate des Modells deutlich verbessert, von 76,41 TP3T auf 90,21 TP3T für AlphaFold 3, was darauf hindeutet, dass bekannte Tascheninformationen die Erfolgsrate der Aufgabe verbessern können. Es gibt jedoch einige Kontroversen über die Definition von Tasche.Wenn Sie also die spezifische Verbesserung von AlphaFold 3 bei der Liganden-Docking-Aufgabe erfahren möchten, können Sie nur die Erfolgsrate des zweiten Aufgabentyps berücksichtigen, der relativ stabiler ist.
Wie in der folgenden Abbildung gezeigt, gibt es bei den Taschendefinitionen zwischen verschiedenen Modellen erhebliche Unterschiede. Die Goldtasche ist eine 25 Å-Kugel (der blaue Teil in der oberen linken Ecke der Abbildung), während das Vina-Modell einen 25 Å-Würfel als Taschendarstellung verwendet, die DeepDock-Taschengröße beträgt 10 Å und die Uni-Mol-Taschengröße beträgt 8 Å.
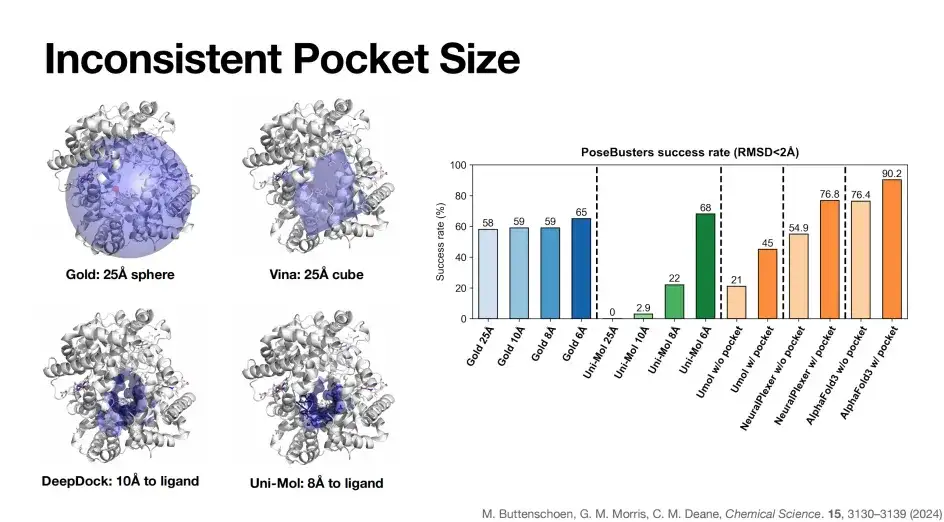
Wie auf der rechten Seite der Abbildung oben gezeigt, ist die Erfolgsrate des PoseBusters-Benchmarks relativ stabil, wenn die Taschengröße des Gold-Modells schrittweise von 25 Å auf 6 Å reduziert wird, was auf die Eigenschaften des physikbasierten Algorithmus von Gold zurückzuführen ist. Im Gegensatz dazu verengte der Deep-Learning-Algorithmus Uni-Mol die Tasche allmählich auf 6 Å, wobei die Erfolgsrate auf 68% anstieg und dann bei 25 Å auf Null sank, was die Abhängigkeit einiger Deep-Learning-Docking-Algorithmen von der Tasche widerspiegelt.
Wie bereits erwähnt, wurde die Docking-Erfolgsrate von AlphaFold 3 nach der Einführung der Tascheninformationen deutlich von 76,4% auf 90,2% verbessert.Zusammenfassend lässt sich sagen, dass Tascheninformationen eine Schlüsselrolle bei der Verbesserung der Erfolgsrate der Modellvorhersage spielen. Idealerweise wäre jedoch ein Modell wie AlphaFold 3 unsere beste Wahl, das eine hohe Genauigkeit ohne Taschen- oder Strukturinformationen erreichen kann.
AlphaFold 3 ermöglicht die Vorhersage von Antikörper- und Antigenstrukturen
Eine weitere Anwendung von AlphaFold 3 ist die Strukturvorhersage von Antikörpern und Antigenen. Auf der linken Seite der Abbildung unten sehen Sie die Leistungsbewertung von AlphaFold 3 für die Vorhersage von Antikörper- und Antigenstrukturen. Bei einem niedrigeren Bewertungsstandard (DockQ > 0,23) liegt die Erfolgsrate von AlphaFold 3 bei nur einem Durchlauf unter 40% (hellblaue Linie), aber nach 1.000 Versuchen kann die Vorhersage-Erfolgsrate auf 60% verbessert werden.
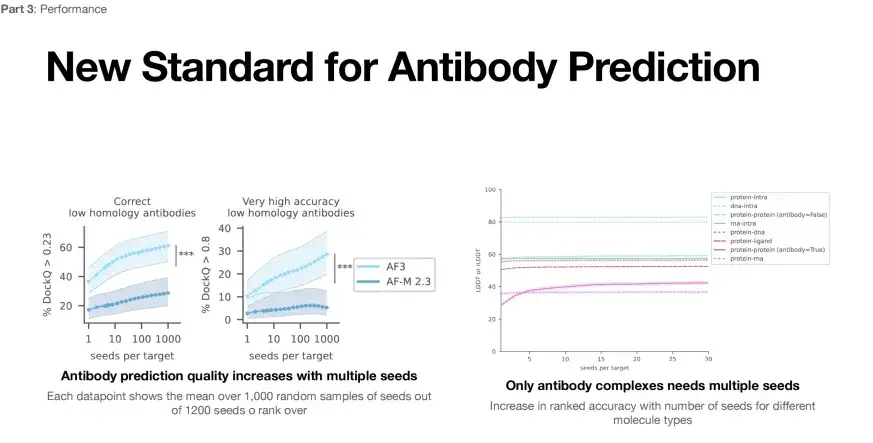
* Links: Vorhersage der Antikörperstruktur, jeder Datenpunkt stellt den Durchschnittswert von 1.000 zufällig aus 1.200 Samen ausgewählten Samen dar
* Rechts: Wenn der Bewertungsindex DockQ größer als 0,23 ist, kann davon ausgegangen werden, dass die strukturelle Genauigkeit noch überprüft werden muss. Wenn DockQ 0,8 überschreitet, ist die Strukturvorhersage sehr genau
Darüber hinaus kann die Erfolgsrate eines einzelnen Durchlaufs bei einer Messung nach einem strengeren Standard (DockQ > 0,8) nur 10% betragen. Durch eine Erhöhung der Anzahl der Durchläufe auf 1.000 kann die Erfolgsrate auf 30% gesteigert werden.Dies zeigt, dass wir die Erfolgsrate der Vorhersage der Antikörper-Antigenstruktur verbessern können, indem wir die Anzahl der AlphaFold 3-Läufe (Seeds pro Ziel) erhöhen.
Wie auf der rechten Seite der obigen Abbildung gezeigt, kann AlphaFold 3 die Erfolgsrate bei der Vorhersage der Struktur von Protein-Protein-Komplexen jedoch nur verbessern, indem die Anzahl der Durchläufe erhöht wird. Dies zeigt, dass die Anwendbarkeit von AlphaFold 3 zur Vorhersage anderer Arten komplexer Strukturen ebenfalls weiter optimiert werden muss.
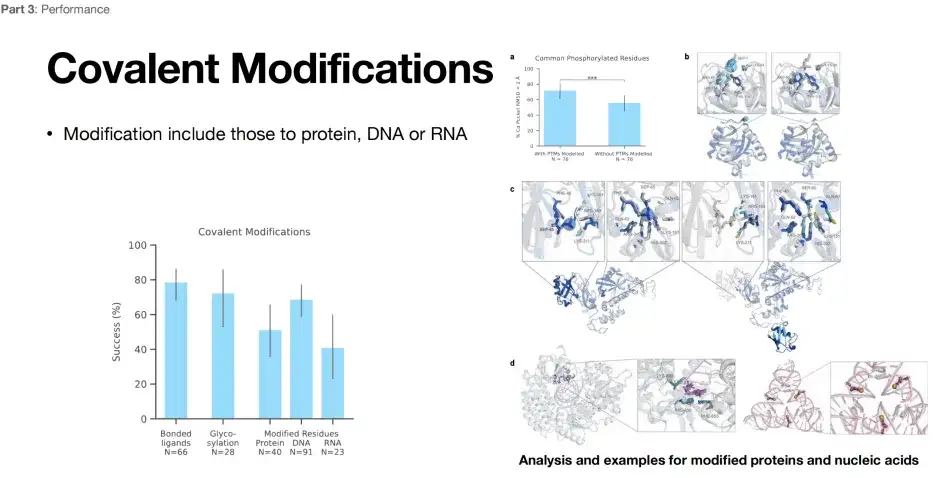
AlphaFold 3 ermöglicht die Vorhersage kovalenter Modifikationen
Wie in der folgenden Abbildung gezeigt, weist AlphaFold 3 auch hervorragende strukturelle Vorhersagefähigkeiten im Hinblick auf die Modifikationsvorhersage auf.Die Erfolgsrate kann etwa 80%, 60% und 40% erreichen. AlphaFold 3 ist zweifellos ein leistungsfähiges Werkzeug für Forscher, die sich mit kovalenten Modifikationen beschäftigen.
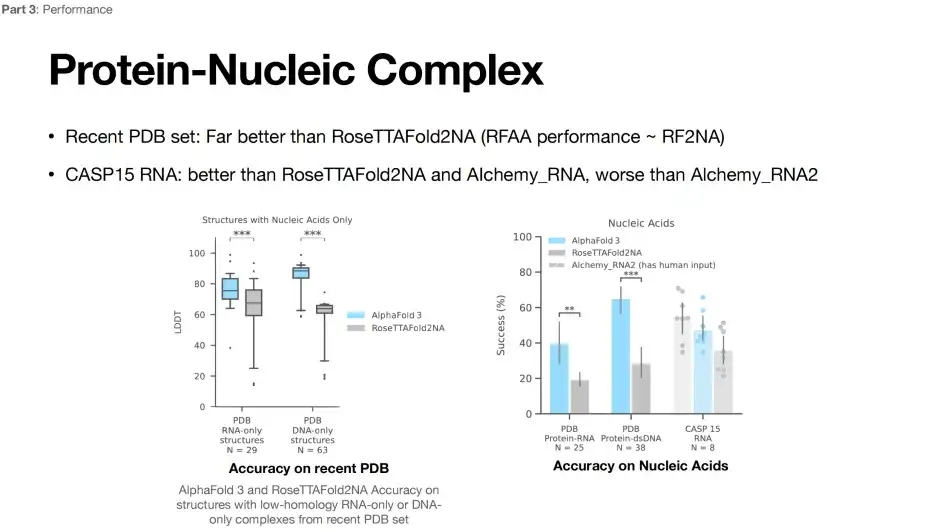
Einschränkungen von AlphaFold 3 bei der Vorhersage von RNA-Strukturen
Derzeit ist die Vorhersage der RNA-Struktur noch schwierig.Wie in der folgenden Abbildung gezeigt, hat AlphaFold 3 die Vorhersageleistung im Vergleich zum RoseTTAFoId2NA-Modell deutlich verbessert. Bei der Vorhersage der CASP15-RNA-Struktur ist die Genauigkeit von AlphaFold 3 jedoch geringer als die des Alchemy_RNA2-Modells (mit menschlichem Input).
Vergleich der Vor- und Nachteile von AlphaFold 3 bei verschiedenen Aufgaben
Durch die Analyse der Trainingskurve von AlphaFold 3 können wir die Leistung des Modells bei verschiedenen Aufgaben deutlich erkennen. Je höher der LDDT-Indikator, desto besser.Wie in der folgenden Abbildung gezeigt, ist das Modell bei der Vorhersage von Intra-Ligandenstrukturen am besten geeignet. es zeigt auch eine hohe Genauigkeit bei der Vorhersage intraproteinaler Strukturen; Dank der stabilen Doppelhelixstruktur der DNA ist das Modell auch bei der Vorhersage intra-DNA-Strukturen gut geeignet. Im Gegensatz dazu ist die Leistung des Modells bei der Vorhersage intra-RNA-Strukturen unzureichend.
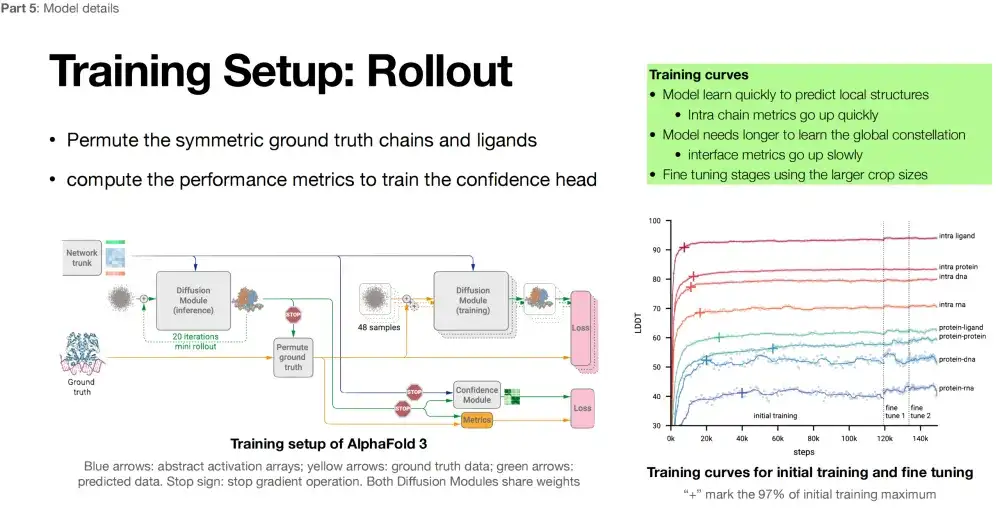
Im Bereich der komplexen Vorhersage schnitt das Modell am besten bei der Vorhersage der Protein-Ligand-Komplexstruktur ab, gefolgt von der Vorhersage von Protein-Protein-Komplexen. Bei der Vorhersage von Protein-DNA-Komplexen nahm die Leistung des Modells ab, und die Vorhersage von Protein-RNA-Komplexen schnitt am schlechtesten ab. Dieses Ergebnis spiegelt auch die Schwierigkeit der Vorhersage der RNA-Struktur wider. Daten zur RNA-Struktur sind rar und die Struktur ist dynamisch und flexibel, was eine der aktuellen Herausforderungen im Bereich der Strukturbiologie darstellt.
Darüber hinaus verwendeten Forscher AlphaFold 3 zur Strukturvorhersage,Die Zuverlässigkeit der Vorhersageergebnisse kann auch anhand der PAE-Tabelle bewertet werden.

AlphaFold 3 ist nicht perfekt
AlphaFold 3 ist nicht perfekt. Beispielsweise kann es sein, dass die falsche Chiralität gewählt wird.Wenn während des Vorgangs eine ungewöhnliche Situation auftritt, wird empfohlen, den Test mehrmals auszuführen, um die Stabilität der Ergebnisse zu überprüfen. Zweitens weist AlphaFold 3 auch Einschränkungen bei der Vorhersage der Proteindynamik auf, was auf den Mangel an Strukturdaten und die Unfähigkeit zurückzuführen sein kann, die mehrdimensionalen Konformationsinformationen von Proteinen zu erfassen.
*Wenn sich ein Objekt von seinem Spiegelbild unterscheidet, wird es als „chiral“ bezeichnet und sein Spiegelbild kann nicht auf das Originalobjekt gelegt werden, genauso wie die linke und die rechte Hand Spiegelbilder voneinander sind und nicht übereinander gelegt werden können.
Darüber hinaus weist AlphaFold 3 auch ein häufiges Problem bei generativen Modellen auf, nämlich Halluzinationen.Wie in den Ergebnissen der Proteinstrukturvorhersage unten gezeigt, kann nur der graue Bereich der Proteinstruktur auf der linken Seite aufgelöst werden, und der Rest befindet sich möglicherweise aufgrund unzureichender Elektronendichte in einem entfalteten Zustand. Das mittlere Bild ist das Ergebnis der Vorhersage des Proteins durch AlphaFold 2. Der blaue Bereich gilt als gefaltet und die anderen „Band“-Teile als entfaltet. Die prognostizierte Struktur ist relativ vernünftig. Rechts ist das Vorhersageergebnis von AlphaFold 3, das dazu neigt, alle möglichen faltbaren Bereiche zu falten. Diese Struktur erscheint sinnvoll, in Wirklichkeit sind die meisten der oben genannten Bereiche jedoch nicht wirklich gefaltet.Daher tendiert die Illusion von AlphaFold 3 dazu, Proteine als gefaltet vorherzusagen, anstatt ihren möglichen entfalteten Zustand beizubehalten.

Um das Halluzinationsproblem von AlphaFold 3 zu lösen,Die Forscher wählten eine direkte und effektive Methode: Da die von AlphaFold 2 vorhergesagten Ergebnisse relativ vernünftig sind, werden die von AlphaFold 2 vorhergesagten Ergebnisse in den Trainingsdatensatz von AlphaFold 3 integriert, um den Trainingseffekt des Modells zu verbessern. Diese Methode weist jedoch eine Einschränkung auf: Wenn bei der Vorhersage von AlphaFold 2 selbst Fehler auftreten, kann dies die Vorhersagequalität von AlphaFold 3 beeinträchtigen, es sei denn, es können andere Datenquellen eingeführt werden, um das Modell weiter zu optimieren.
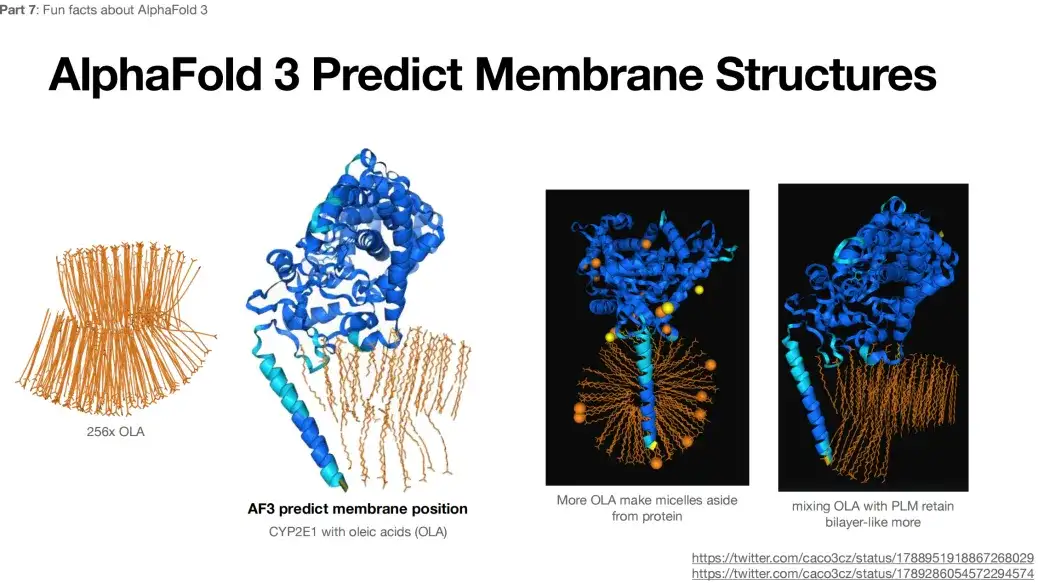
Wenn außerdem 256x OLA als Eingabe an AlphaFold 3 übermittelt wird, zeigt das vorhergesagte Ergebnis eine doppelschichtähnliche Struktur, wie in der folgenden Abbildung dargestellt.Diese Struktur ist weder zu erwarten noch typisch.
Darüber hinaus ist AlphaFold 3 bei der Vorhersage der Struktur von RNA und DNA nicht genau.Wie in der Abbildung unten gezeigt, treten bei der Vorhersage der RNA-Struktur sogar bizarre komplementäre Paarungen auf, wie etwa G:G, G:A usw.
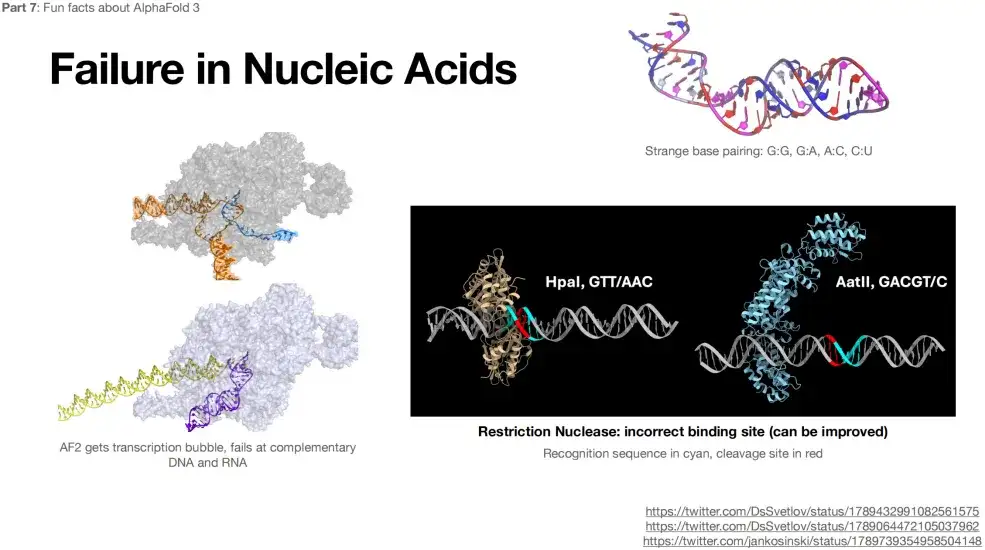
Einschränkungen bei der Verwendung von AlphaFold 3
Unter der Voraussetzung, dass die Daten nicht streng vertraulich sind, kann jeder über die von Google bereitgestellte Website auf AlphaFold 3 zugreifen. Allerdings weist die Plattform auch einige Einschränkungen auf. Wie in der folgenden Abbildung gezeigt, unterstützt AlphaFold 3 im Hinblick auf die Proteinmodifikation derzeit nur eine begrenzte Anzahl von Modifikationen an 3 bestimmten Positionen, insgesamt 23 Typen. Die DNA-Modifikation unterstützt nur 9 Typen, die RNA-Modifikation nur 15 Typen, Metallionen unterstützen nur 10 verschiedene Metalle und Liganden sind auf 14 kleine Moleküle beschränkt.
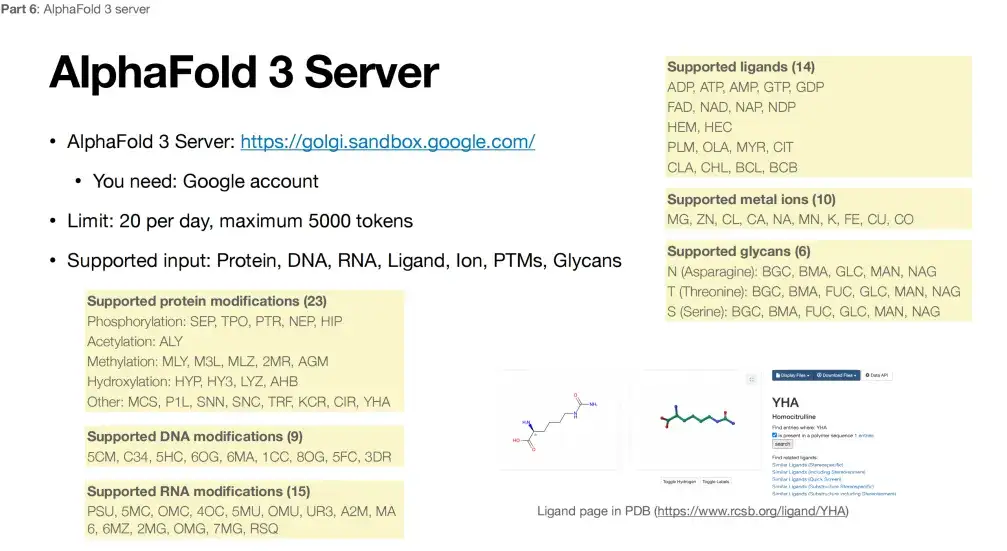
Angesichts der oben genannten spezifischen Einschränkungen ist AlphaFold 3 möglicherweise nicht in der Lage, die meisten Forschungsarbeiten und Reaktionen zu verarbeiten und muss möglicherweise warten, bis es wirklich Open Source ist.
Zusammenfassend lässt sich sagen, dass AlphaFold 3 bei der Erweiterung seines Vorhersageumfangs bedeutende Erfolge erzielt und bestehende KI-Modelle übertroffen hat. Seine Leistung bei bestimmten Aufgaben muss jedoch noch verbessert werden, insbesondere bei der Vorhersage feiner Strukturen.Obwohl bei AlphaFold 3 erhebliche Fortschritte erzielt wurden, sind daher weiterhin weitere Forschung und Anstrengungen erforderlich, um bestimmte komplexe Probleme vollständig zu lösen.
Über Zhong Bo Zitao

Zhong Bozitao ist derzeit Doktorand für künstliche Intelligenz an der Shanghai Jiao Tong University. Zu seinen wichtigsten Forschungsschwerpunkten gehören die Hochdurchsatzvorhersage von Proteinstrukturen und -funktionen, die Generierung von Proteinkonformationen usw. Seit 2019 hat er über 20 Artikel veröffentlicht und die Ergebnisse der Hochdurchsatzanalyse zur AlphaFold-Strukturvorhersage der Beziehung zwischen Tiefseeproteom und Stoffwechselwegen in Nature Communications veröffentlicht. Er hat dreimal die Goldmedaille der International Genetically Engineered Machine Competition (iGEM) gewonnen und war viele Male als Juror für den Wettbewerb tätig.
Google Scholar:
https://scholar.google.com/cita








