Command Palette
Search for a command to run...
KI Für Die Genomik | Algorithmus Zur Räumlichen Transkriptomdatendarstellung SPACE, Anwendung Künstlicher Intelligenz in Der Genomik

In der zweiten Folge der Serie „Meet AI4S“ haben wir die Ehre, Li Yuzhe, einen Postdoktoranden im Labor von Zhang Qiangfeng an der School of Life Sciences der Tsinghua-Universität, einzuladen.Sein Labor, Zhang Qiangfeng, gehört zur School of Life Sciences der Tsinghua-Universität. Es ist außerdem ein wichtiger Teil des gemeinsamen Zentrums für Biowissenschaften der Tsinghua-Peking-Universität und des Beijing Advanced Innovation Center for Structural Biology. Die Forschung des Labors konzentriert sich auf die Schnittstelle zwischen Biowissenschaften und Algorithmen der künstlichen Intelligenz, auf Technologie und Algorithmenentwicklung für RNA-Strukturgruppen, Technologie und Algorithmenentwicklung für die Sequenzierung einzelner Zellgenome, Modellierung von Proteinstrukturen auf der Grundlage von Daten der Kryo-Elektronenmikroskopie und die Entwicklung damit verbundener Algorithmen für künstliche Intelligenz.
Dieses Teilen,Dr. Li Yuzhe hielt eine Rede mit dem Titel „Erforschung von KI-Anwendungen in der Genomik: Am Beispiel des Algorithmus zur räumlichen Transkriptomdatencharakterisierung SPACE“.Die neuesten Forschungsergebnisse des Teams wurden geteilt und KI-Methoden in der räumlichen Transkriptomik und Einzelzell-Omics-Forschung vorgestellt.
HyperAI hat die ausführlichen Ausführungen von Dr. Li Yuzhe zusammengestellt und zusammengefasst, ohne die ursprüngliche Absicht zu verletzen.
Klicken Sie hier, um die vollständige Live-Wiederholung anzuzeigen:
KI für die Wissenschaft bringt enorme Veränderungen im Forschungsparadigma im wissenschaftlichen Bereich mit sich
Heute möchte ich über das Thema „KI für die Wissenschaft“ sprechen. Ich bin davon überzeugt, dass KI für die Wissenschaft zu großen Veränderungen im Forschungsparadigma im gesamten wissenschaftlichen Bereich geführt hat. Im Folgenden werde ich dies am Beispiel der Proteinstrukturforschung genauer erläutern.
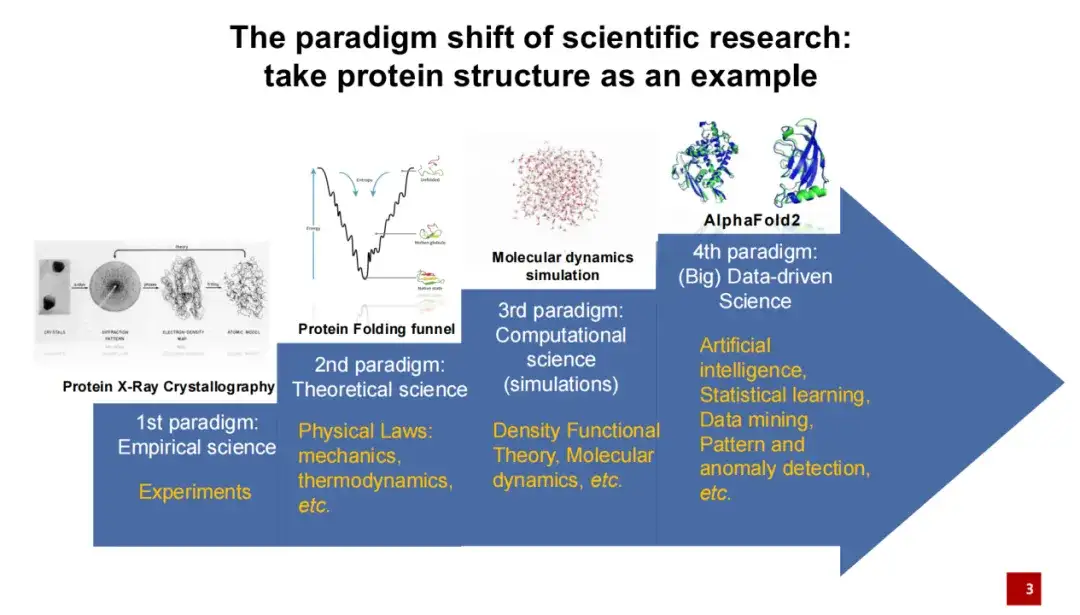
Die erste Generation des Paradigmas der Proteinstrukturforschung wurde hauptsächlich mit experimentellen Mitteln durchgeführt.Das heißt, man fotografiert die vom Protein gebildeten Kristalle mit Röntgenstrahlen und führt dann eine Strukturmodellierung durch.
Die zweite Generation des Proteinstrukturforschungsparadigmas wurde hauptsächlich von Physikern angeführt, die der Untersuchung der Proteinstruktur theoretisches Wissen hinzufügten.Wenn beispielsweise die Energie einer Proteinfaltung gering ist, dann ist diese Faltung relativ stabil.
Die dritte Generation des Paradigmas der Proteinstrukturforschung bezieht sich auf die 1990er Jahre, als mit der Entwicklung der Computertechnologie die Computersimulation schrittweise in der Proteinstrukturforschung eingesetzt wurde.Insbesondere molekulardynamische Simulationen haben in den letzten Jahren große Verbreitung gefunden. Diese Simulationsmethoden helfen uns, Proteinstrukturen bis zu einem gewissen Grad besser zu berechnen und vorherzusagen. In den letzten Jahren, insbesondere im Jahr 2020, haben Algorithmen der künstlichen Intelligenz Einzug in das Gebiet der Proteinstruktur gehalten und einen weiteren Durchbruch bewirkt. Beim Wettbewerb zur Vorhersage von Proteinstrukturen 2020 war AlphaFold 2 anderen konkurrierenden Methoden weit voraus.
Die Einführung künstlicher Intelligenz hat einen enormen Paradigmenwechsel in den Biowissenschaften und im gesamten Bereich der wissenschaftlichen Forschung bewirkt. Im Vergleich zu traditionellen Forschungsmethoden,Bei der künstlichen Intelligenz wird stärker Wert darauf gelegt, von Daten auszugehen und datengesteuerte wissenschaftliche Forschung zu betreiben.Dies bedeutet, dass wir nicht mehr im Voraus eine wissenschaftliche Hypothese aufstellen müssen, sondern die Naturgesetze direkt aus den Daten lernen und aufdecken können.
Die Entwicklung der KI für die Genomik
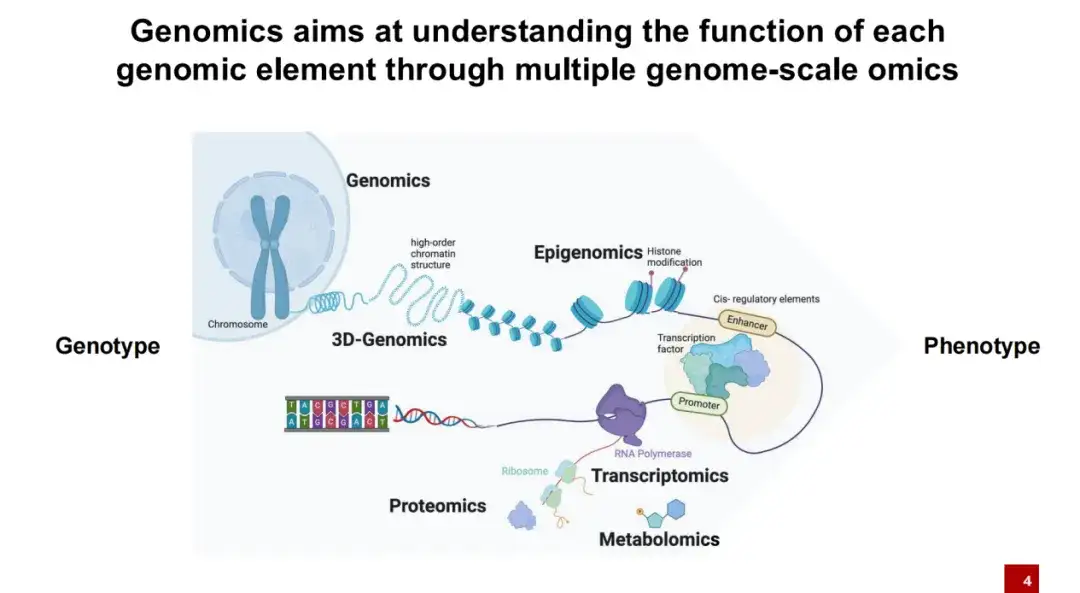
Der folgende Austausch konzentriert sich auf die Anwendung von KI im Bereich der Genomik. Zusamenfassend,Die Genomforschung untersucht hauptsächlich die Beziehung zwischen Genotyp (die gesamte DNA im Körper) und Phänotyp (individuelle Merkmale wie Größe und Gewicht).
Wie wir alle wissen, liegt die DNA in Zellen nicht nackt vor, sondern ist um Nukleosomen gewickelt. Nukleosomen sind mit vielen Histonmodifikationen verbunden. Normalerweise sind diese DNAs eng miteinander verwoben. Nur unter bestimmten Bedingungen wird die DNA freigelegt und es bildet sich ein offenes Intervall. An diesem Punkt können Proteine wie Transkriptionsfaktoren an diese freiliegenden DNA-Regionen binden.
Im anschließenden Transkriptionsprozess kann RNA durch RNA-Polymerase transkribiert und dann durch Ribosomen in Protein übersetzt werden, und das Protein spielt schließlich eine Rolle bei Lebensaktivitäten.Das Forschungsziel der Genomik besteht darin, zu verstehen, wie verschiedene DNA-Elemente die Lebensaktivitäten beeinflussen.
Wir fassen die wichtigen Ereignisse und Entwicklungen in der Entwicklung der KI für die Wissenschaft seit der Entschlüsselung der DNA-Doppelhelixstruktur in den 1950er Jahren bis in die jüngste Vergangenheit zusammen. Seine Ursprünge lassen sich auf die Entdeckung der Doppelhelixstruktur der DNA in den 1950er Jahren und die Entwicklung der Sanger-Sequenzierungstechnologie in den 1970er Jahren zurückführen.
Wie in der Abbildung unten gezeigt, stellt der blaue Teil die Entwicklung verschiedener Sequenzierungstechnologien und experimenteller Technologien dar; der grüne Teil stellt wichtige Methoden im Bereich der künstlichen Intelligenz dar; der gelbe Teil stellt die Einrichtung einiger wichtiger groß angelegter Forschungspläne und Datenbanken dar; Der rote Teil stellt repräsentative Methoden und Anwendungen im Bereich der KI für die Genomik dar.
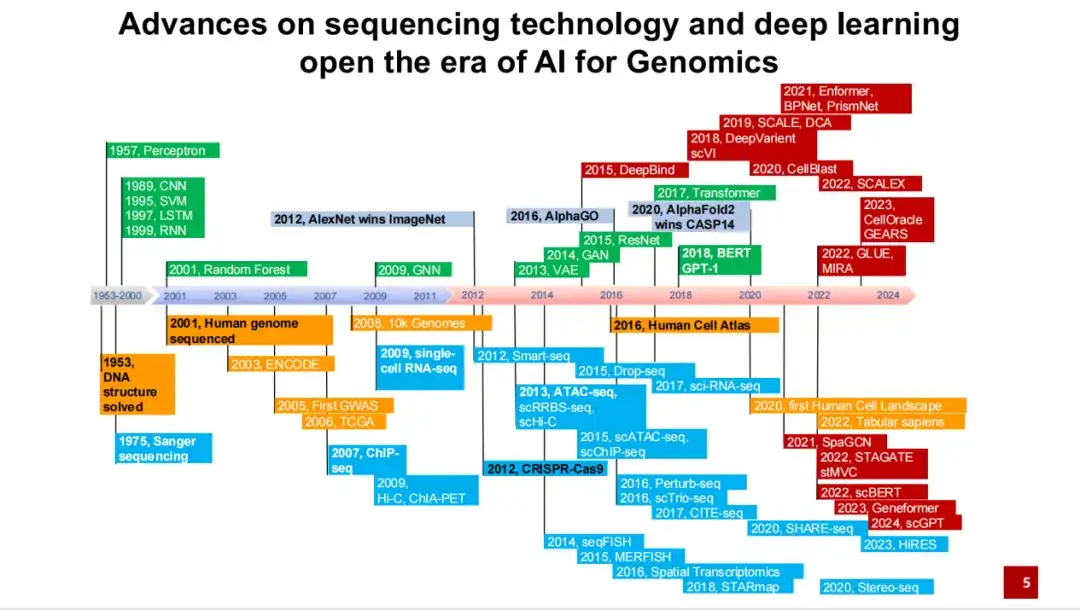
Wie Sie sehen können,2001wurde der Entwurf des Humangenomprojekts zunächst fertiggestellt, wobei die gesamte DNA-Sequenz eines weißen Mannes sequenziert wurde. Im Jahr 2012AlexNet übertraf zum ersten Mal den Menschen bei Bildklassifizierungsaufgaben und leitete damit die explosive Entwicklung der künstlichen Intelligenz im letzten Jahrzehnt ein. Im Jahr 2016Das Projekt „Human Cell Atlas“ wurde vorgeschlagen und die Forschung verlagerte sich allmählich von der DNA-Sequenz eines einzelnen Individuums auf alle Zellen. Im selben Jahr besiegte AlphaGo, basierend auf Methoden des bestärkenden Lernens, die Menschen im Go-Spiel.
Für KI für die Genomik oder KI für die Wissenschaft,Ein wichtiger Durchbruch war, dass AlphaFold 2 im Jahr 2020 bei CASP 14 einen mit großem Abstand ersten Platz belegte.Dies hat dazu geführt, dass immer mehr Methoden der künstlichen Intelligenz im Bereich der Genomik eingesetzt werden.
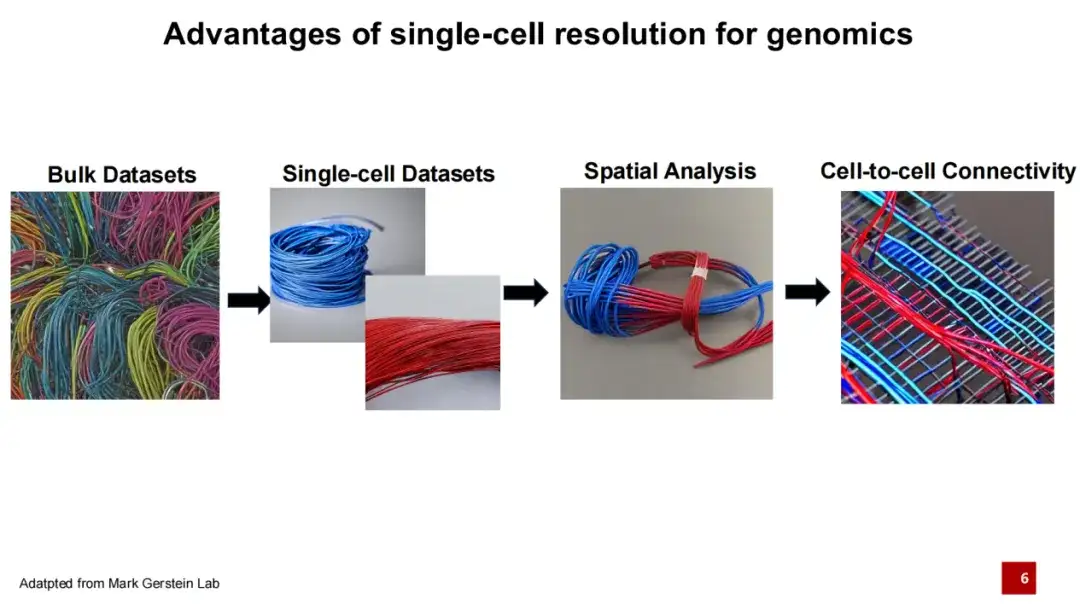
In,Die Einzelzellgenomik stellt einen bedeutenden Durchbruch auf dem Gebiet der Genomik der letzten Jahre dar.Bei der traditionellen Genomforschung handelt es sich üblicherweise um Massensequenzierung. Gehen Sie davon aus, dass jede Linie in der folgenden Abbildung einen Zelltyp darstellt und Linien unterschiedlicher Farbe unterschiedliche Zelltypen darstellen. Bei früheren Sequenzierungsmethoden musste das gesamte Gewebe gemischt und sequenziert werden, wodurch es schwierig war, festzustellen, aus welcher spezifischen Zelle jede DNA oder RNA stammte. Durch die Entwicklung der Einzelzelltechnologie können wir nicht nur die gesamte DNA oder RNA in einem Gewebe gewinnen, sondern auch die spezifische zelluläre Quelle dieser DNA oder RNA identifizieren. Da unterschiedliche Zelltypen unterschiedliche Genexpressionen aufweisen und unterschiedliche Funktionen erfüllen, können wir die Lebensaktivitäten besser verstehen.
In den letzten fünf Jahren hat die räumliche Omics-Technologie, repräsentiert durch die räumliche Transkriptomik, auf der Grundlage der Einzelzell-Omics-Technologie einen Schritt weitergegangen.Wir können nicht nur Informationen über jeden Zelltyp erhalten, sondern auch die Verteilung dieser Zellen im Raum bestimmen.Da Interaktionen zwischen Zellen eine wichtige Grundlage für die Realisierung ihrer Funktionen darstellen, konzentriert sich die weitere Forschung auf die Frage, wie Zellen miteinander verbunden sind.
Seit dem Start des Humangenomprojekts bis zum Vorschlag des Human Cell Atlas-Projekts im Jahr 2016 bestand sein Ziel darin, die Referenzkarte aller menschlichen Zellen zu vervollständigen, um uns zu einem besseren Verständnis der Lebensaktivitäten zu verhelfen und Unterstützung bei der Behandlung und Diagnose bestimmter Krankheiten zu bieten.
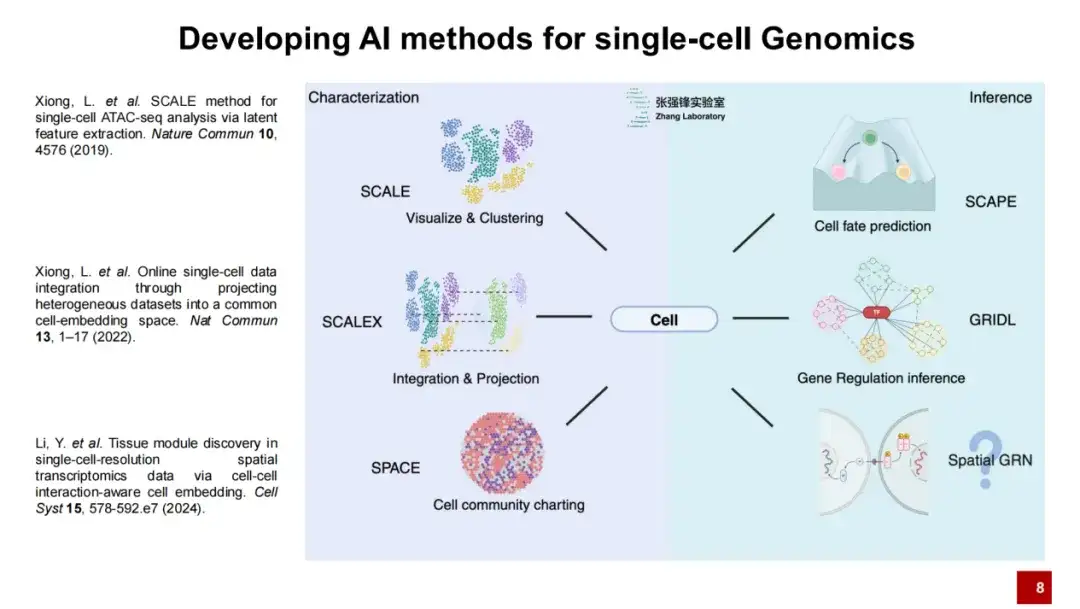
Das Team entwickelte drei Methoden: SCALE, SCALEX und SPACE für die Einzelzellgenomikforschung
Unser Labor hat eine Reihe von Methoden der künstlichen Intelligenz entwickelt.Wir glauben, dass die Einzelzellgenomik zwei wichtige Schritte erfordert: erstens die Beschreibung der Zelle und zweitens die Schlussfolgerung über die Zelle.
Wir haben drei Arbeiten zur Beschreibung von Zellen veröffentlicht: SCALE, SCALEX und SPACE.SCALE dient hauptsächlich der Visualisierung und Clusterbildung, SCALEX der Datenintegration und -projektion und SPACE der Beschreibung der Mikroumgebung der gesamten räumlichen Transkriptomdatenorganisation.Heute werde ich hauptsächlich die beiden Methoden vorstellen: SCALEX und SPACE.
SCALEX-Methode zur Eliminierung von Batch-Effekten
Das SCALEX-Verfahren dient der Eliminierung von Batch-Effekten.Dies ist ein sehr wichtiges Thema in der Genomforschung. Unter Batch-Effekt versteht man Unterschiede in den Versuchsergebnissen verschiedener Chargen, die auf technische Faktoren wie unterschiedliche Versuchsbedingungen zurückzuführen sind.
Wie in der Abbildung unten gezeigt, sollte die Sequenzierung dieser beiden Zellgruppen theoretisch eine sehr ähnliche Genexpression ergeben, selbst wenn wir zwei biologische Replikate von Zellen separat kultivieren. Aus technischen Gründen, wie z. B. Unterschieden in der Kulturumgebung, der Bibliotheksaufbauzeit oder der Sequenzierungsplattform,Die endgültigen Genexpressionsprofile können stark variieren und daher viel technisches Rauschen verursachen.Daher ist es bei der Analyse der Daten notwendig, diesen Batch-Effekt zu entfernen.

In der biologischen Forschung können Daten oft nicht auf einmal erhoben werden, sondern müssen nach und nach durch mehrere Experimente angesammelt werden. daher,Entfernen Sie Batch-Effekte und analysieren Sie Daten auf integrierte Weise, um Faktoren zu finden, die wirklich biologisch relevant sind.Es handelt sich um einen entscheidenden Schritt in der Genomforschung bzw. Einzelzellgenomikforschung.
Darauf aufbauend haben wir die SCALEX-Methode entwickelt.Es ist in der Lage, die verarbeiteten Einzelzelldaten in einen verallgemeinerten zellulären latenten Raum zu projizieren. Das Framework von SCALEX basiert auf Variational Autoencodern (VAE).
Der erste Input sind die Transkriptomdaten einer einzelnen Zelle, die dann durch einen batchfreien Encoder in einen verallgemeinerten zellulären latenten Raum projiziert werden.
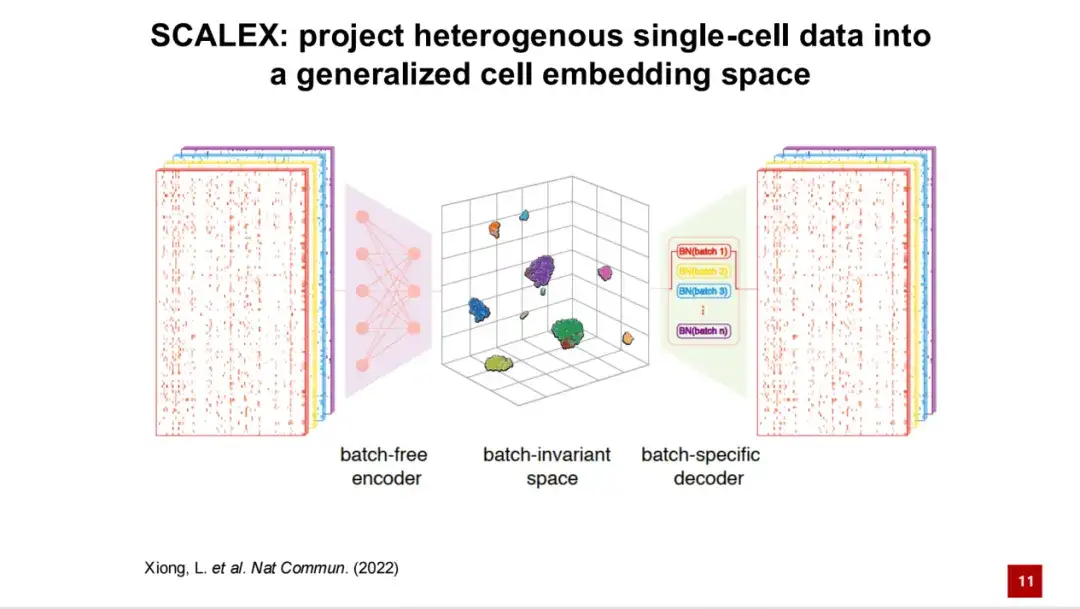
Anschließend werden die Batch-Informationen durch domänenspezifische Batch-Normalisierung über einen Batch-spezifischen Decoder zum Modell hinzugefügt. Durch dieses asymmetrische Design ist der generierte zelllatente Raum ein chargenunabhängiger Raum, der theoretisch kein chargenbezogenes technisches Rauschen enthält. Die Genexpression wird durch den Decoder rekonstruiert und der Verlust wird mit dem ursprünglichen Eingabe-Genexpressionsspektrum berechnet. Gleichzeitig wird in Kombination mit der KL-Divergenz die Verlustfunktion des SCALEX-Modells konstruiert, bei dem es sich um ein selbstüberwachtes Modell handelt.
Dieses asymmetrische Encoder- und Decoder-Design hat zwei Hauptvorteile:Erstens ist der resultierende Encoder universell.Das heißt, neue Daten können ohne Batch-Informationen direkt durch den Encoder in den latenten Zellraum projiziert werden, ohne dass das Modell neu trainiert oder die neuen Daten erneut in die vorhandenen Daten integriert werden müssen.
Zweitens schenkt SCALEX dem globalen Batch-Effekt mehr Aufmerksamkeit.Die traditionelle Methode zum Entfernen von Batch-Effekten besteht hauptsächlich darin, ähnliche Zellen (Zellenpaare) in zwei Datenstapeln zu finden und sie zur Korrektur zu paaren, um den Batch-Effekt zu entfernen. Bei dieser Methode handelt es sich im Wesentlichen um eine relativ lokale Batch-Effektkorrektur.
Bei dieser Art von Methode gibt es jedoch ein Problem: Bei der tatsächlichen Datenanalyse sind die Zelltypen in zwei verschiedenen Chargen möglicherweise nicht vollständig konsistent, es gibt möglicherweise nur wenige gemeinsame Zelltypen und der Rest ist chargenspezifisch. Wenn die Zellpaarung erzwungen wird, kann es zu einer Überkorrektur kommen, weil keine geeigneten gepaarten Zellen gefunden werden können und Zelltypen, die nicht ausgerichtet werden sollten, zur Ausrichtung gezwungen werden.
In diesem Zusammenhang werde ich die beiden wesentlichen Vorteile von SCALEX genauer erläutern.
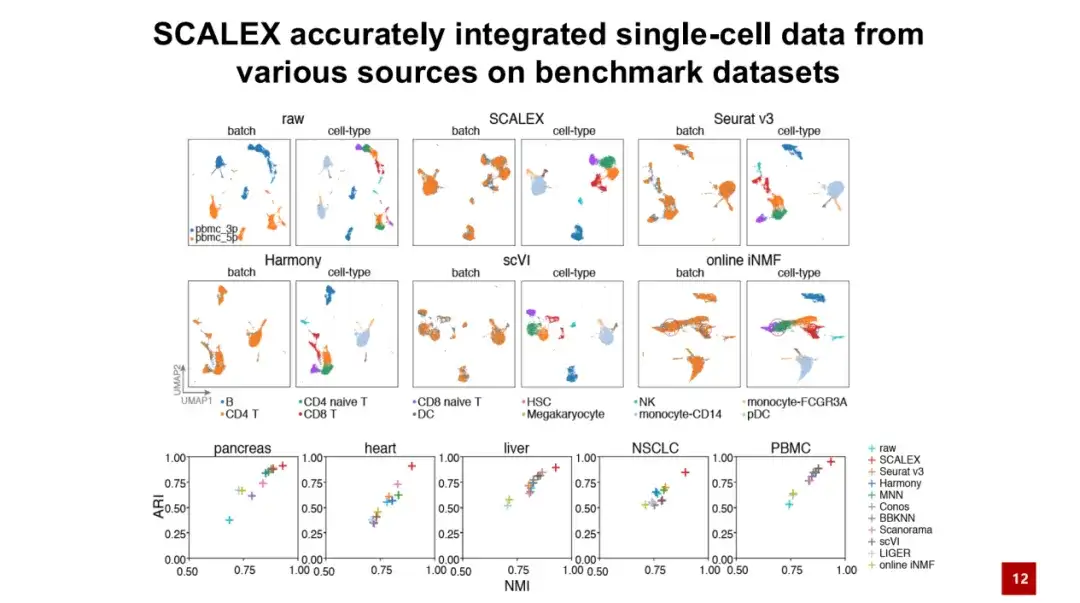
Zunächst führen wir ein Benchmarking von SCALEX anhand von fünf Testdatensätzen durch.Die Ergebnisse zeigen, dass SCALEX hinsichtlich der Genauigkeit bestehende Methoden übertrifft.
Wie in der folgenden Abbildung gezeigt, stellt das Batch-Diagramm die ursprünglichen und unkorrigierten Daten dar, Blau und Orange stellen jeweils zwei Datenbatches dar und der Zelltyp stellt den Zelltyp dar. Es ist ersichtlich, dass diese beiden Chargen zwar ähnliche Zelltypen enthalten, aufgrund des großen Chargeneffekts jedoch Zellen, die ursprünglich zum selben Zelltyp gehörten, nicht zusammengeführt werden konnten. Dies führte dazu, dass technische Faktoren biologische Faktoren verdeckten und nachfolgende biologische Forschung unmöglich machten.
Nach der SCALEX-Integration konnten die beiden Zellchargen gut aggregieren und waren klar nach Zelltypen getrennt, was die Bedeutung von SCALEX in praktischen Anwendungen verdeutlicht.
Ein wichtiger Vorteil von SCALEX besteht darin, dass es zwei Datenstapel mit denselben Zelltypen verarbeiten kann.Solche Daten werden als teilweise überlappende Datensätze bezeichnet. Wie in der folgenden Abbildung gezeigt, bedeutet Überlappung 0, dass die Zelltypen in den beiden Stapeln völlig unterschiedlich sind, und Überlappung 4 bedeutet, dass in den beiden Stapeln 4 gemeinsame Zelltypen vorhanden sind.
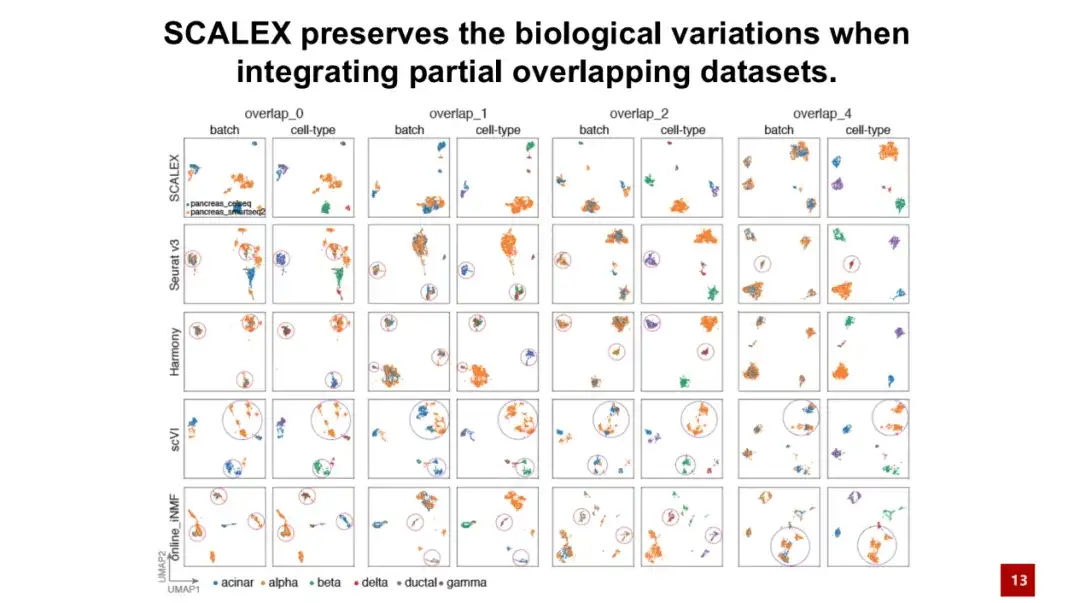
Die Ergebnisse zeigen, dass SCALEX auch dann biologische Unterschiede gut aufrechterhalten kann, wenn die beiden Zellchargen überhaupt keine identischen Zelltypen aufweisen. Das heißt, SCALEX integriert Zellen unterschiedlichen Zelltyps nicht zwangsweise zusammen, während andere ähnliche Methoden auf der Suche nach Zellpaarungen beruhen können, was zu einer Überkorrektur führt.
Ein weiterer Vorteil von SCALEX besteht darin, dass der universelle Encoder neue Daten ohne Batch-Effekte direkt in den vorhandenen latenten Raum der Zellen projizieren kann, ohne das Modell neu trainieren zu müssen.Wie in der folgenden Abbildung gezeigt, wird zunächst mithilfe der Pankreas-Datensätze ein Referenzzellatlas trainiert. Anschließend werden die drei neuen Daten durch den trainierten Encoder direkt in den latenten Zellraum projiziert. Die Farben in der Abbildung stellen Zelltypen dar und die grauen Punkte stellen die konstruierten Referenzzelltypen dar. Es lässt sich feststellen, dass die verschiedenen Zelltypen in ihren jeweiligen Positionen in der Abbildung gut voneinander getrennt sind.
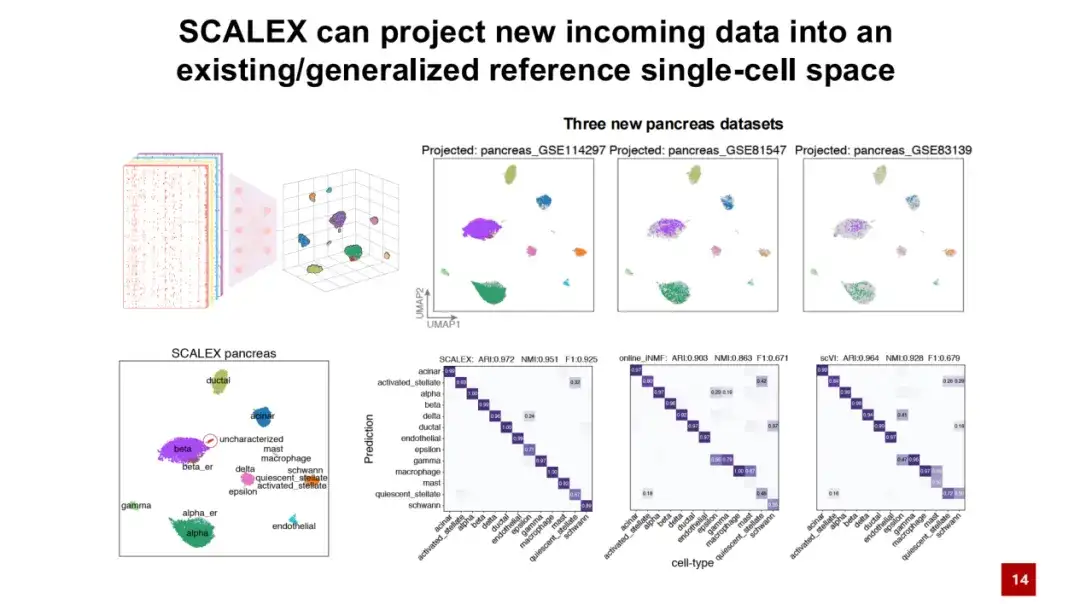
Indem wir die Beschriftungen von Referenzzellen um eine bestimmte Position herum auf neue Datenzellen projizieren, können wir sehen, dass SCALEX bei der automatischen Annotation von Zelltypen gute Ergebnisse erzielt. Im Vergleich zu anderen bestehenden Methoden hat SCALEX eine sehr wichtige Anwendung.Das heißt, neue Daten können direkt in die erstellten Daten projiziert werden, was uns dabei hilft, vergleichende Analysen zwischen den Daten durchzuführen.
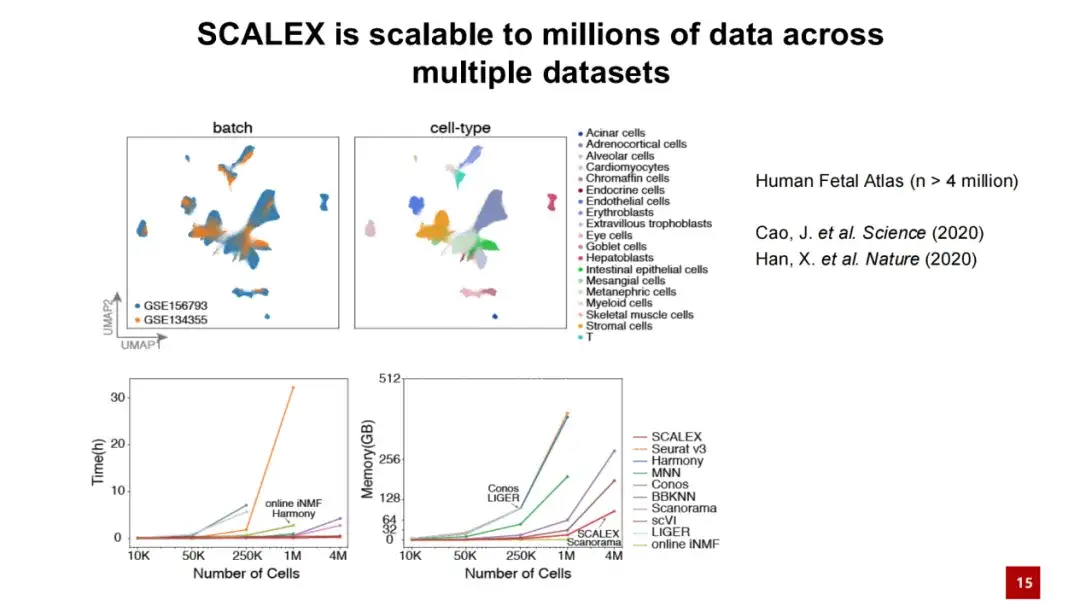
Darüber hinaus ist SCALEX auch bei der Verarbeitung großer Datenmengen leistungsstark. Die folgende Abbildung zeigt, dass bei der Verarbeitung von 4 Millionen Zelldaten durch SCALEX die Berechnungszeit einige zehn Minuten nicht überschreitet und der Speicherverbrauch weniger als 100 GB beträgt. Dies zeigt, dass SCALEX eine gute Skalierbarkeit aufweist und für die integrierte Analyse von Einzelzelldaten im extrem großen Maßstab verwendet werden kann.
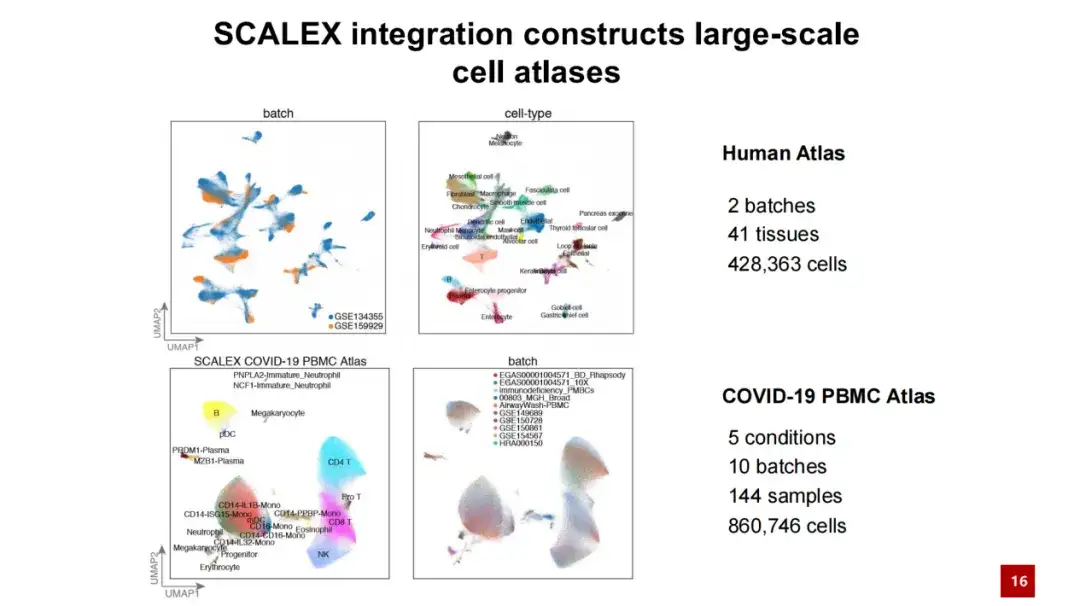
Unter Ausnutzung der Vorteile von SCALEX haben wir zwei groß angelegte Zellatlanten erstellt: einen Zellatlas menschlicher Individuen mit mehr als 400.000 Zellen; der andere ist ein COVID-19-PBMC-Zellatlas, der mehr als 860.000 Zellen und mehr als 100 Proben enthält.
SPACE: ein KI-Analysetool für räumliche Transkriptomdaten
Als Nächstes werde ich Ihnen das räumliche Transkriptomanalysetool SPACE vorstellen, das mein Team kürzlich veröffentlicht hat.
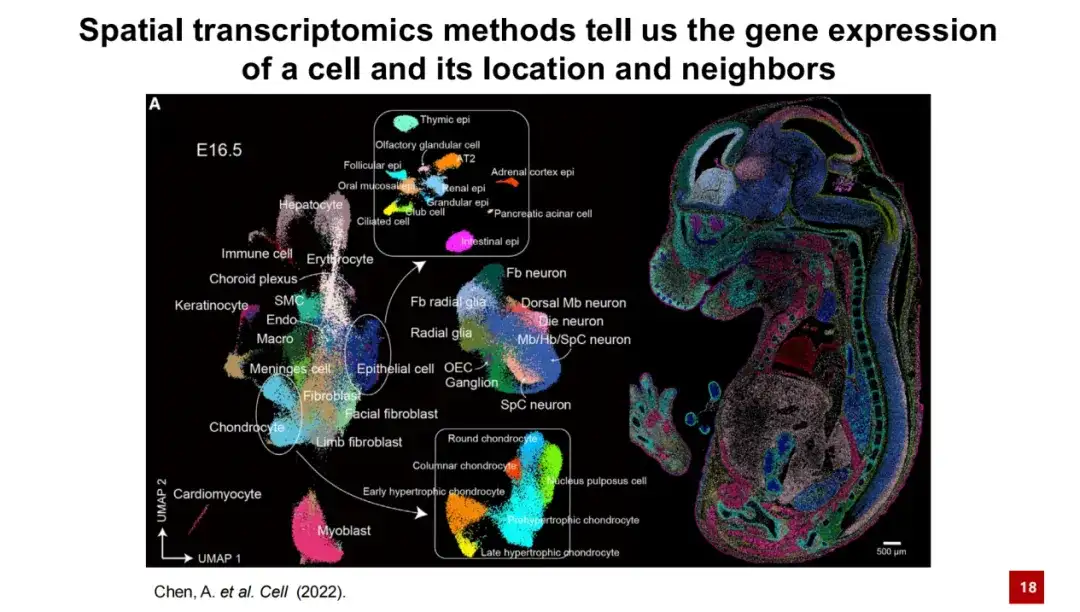
Einfach ausgedrückt kann die räumliche Transkriptomik-Technologie Informationen zur Genexpression von Zellen und ihrer spezifischen Position im Raum liefern. Die folgende Abbildung zeigt ein typisches räumliches Transkriptomergebnis. In der linken Abbildung stellt jeder Punkt eine Zelle dar und die Farbe gibt ihren Zelltyp an. Diese Zellen wurden durch Dimensionsreduktion der Genexpression gruppiert, um eine UMAP-Karte zu bilden. Im rechten Bereich wird die tatsächliche räumliche Position jeder Zelle in den Daten des Mausembryos E16.5 angezeigt. Es ist deutlich zu erkennen, dass die räumliche Verteilung der Zellen eine gute Spezifität aufweist.
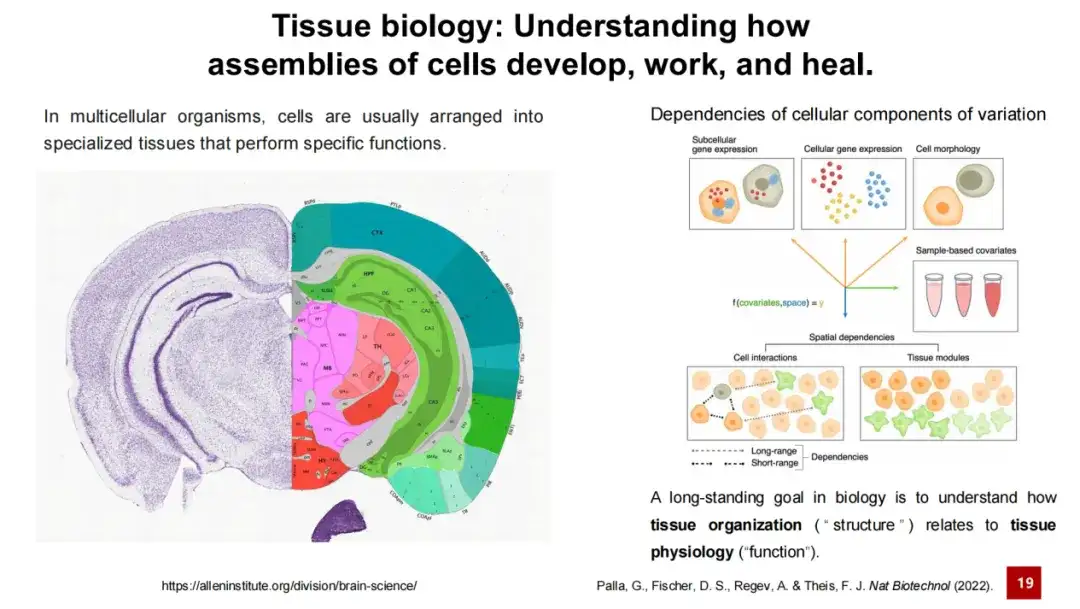
Die Organisationsforschung ist seit jeher eines der Kernthemen der lebenswissenschaftlichen Forschung. Man kann sagen, dass eines der langfristigen Ziele der biologischen Forschung darin besteht, die Beziehung zwischen der Struktur einer Organisation und ihrer Funktion zu verstehen. Das ist leicht zu verstehen. Beispielsweise bestehen verschiedene Gehirnregionen aus unterschiedlichen Neuronen und Stützzellen, die durch komplexe Zell-Zell-Interaktionen unterschiedliche Funktionen erfüllen. Beispielsweise sind einige Bereiche für das Gedächtnis verantwortlich, andere für das Lernen und wieder andere für motorische Reaktionen.
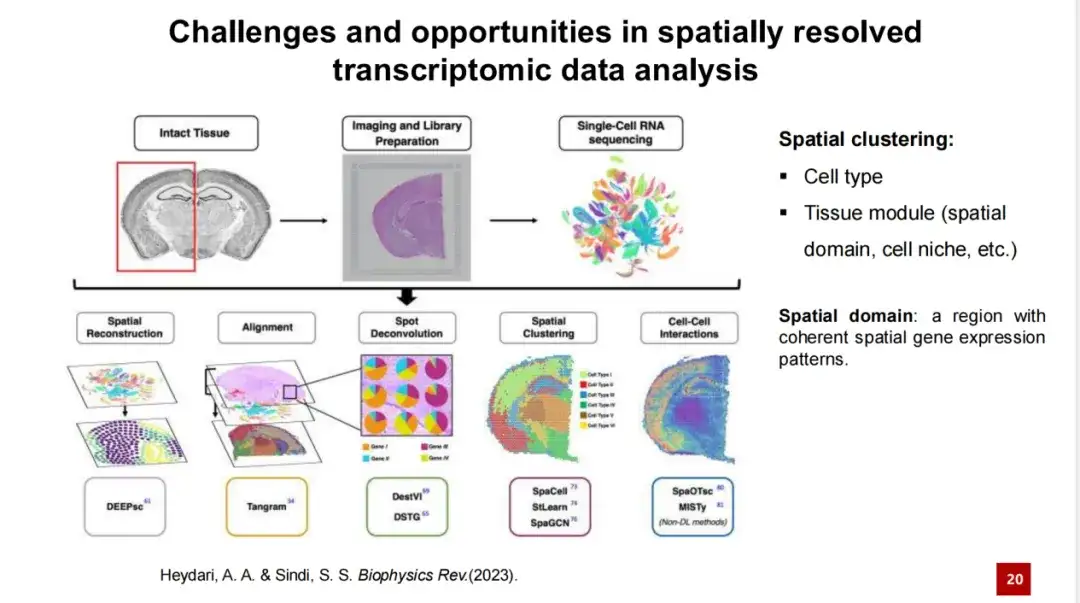
daher,Ein Kernproblem bei der räumlichen Transkriptomanalyse besteht darin, verschiedene Zelltypen oder Gewebemodule im Raum zu identifizieren. Diese Aufgabe wird zusammenfassend als räumliche Clusterung bezeichnet.
Diese Aufgabe besteht aus zwei Teilaufgaben: Die eine besteht in der Identifizierung des Zelltyps und die andere in der Identifizierung des Gewebemoduls.. Ersteres ist intuitiver, nämlich die Identifizierung verschiedener Zelltypen in räumlichen Transkriptomdaten, wie sie in Daten zu Mausembryos gezeigt werden; Letzteres ist relativ abstrakt und beinhaltet die Identifizierung von Bereichen innerhalb des Gewebes, die kleiner als die Gewebestruktur sind und bestimmte Funktionen haben oder aus Zellen bestehen können.
In verschiedenen Studien geben Forscher Gewebemodulen unterschiedliche Namen, beispielsweise räumliche Domäne oder Zellnische, wobei räumliche Domäne der gebräuchlichste Begriff ist. Einige Forscher glauben, dass es bei der Identifizierung von Organisationsmodulen darum geht, Regionen mit konsistenten räumlichen Genexpressionsmerkmalen zu identifizieren.
Dieses Konzept weist jedoch Einschränkungen auf. Abbildung A unten zeigt beispielsweise, dass zwischen zwei Regionen ein signifikanter Unterschied in der Genexpression besteht, in den Abbildungen B und C ist die Verteilung der Genexpression zwischen den Regionen jedoch nicht ganz klar und kann verfälscht sein. Die Abbildungen B und C zeigen Situationen, die mit dem räumlichen Domänenkonzept nicht gelöst werden können.

Um dieses Problem zu lösen, schlagen wir die SPACE-Methode vor.Das Problem des räumlichen Bereichs wird durch das Erlernen interaktionsbewusster Zelleneinbettungen angegangen.
SPACE verwendet ein Graph-Autoencoder-Framework, um niedrigdimensionale Zelleinbettungen zu lernen.
Zuerst geben wir die räumlichen Transkriptomdaten ein und erstellen einen Nachbargraphen basierend auf der räumlichen Position jeder Zelle, d. h., wir verbinden die nächstgelegenen Nachbarzellen jeder Zelle, um einen Graphen zu bilden. In der folgenden Abbildung stellen Knoten Zellen dar und die Eigenschaften der Knoten sind die Genexpressionsmerkmale der Zellen. Wir geben die benachbarten Graphen und Genexpressionsprofile in den Encoder von SPACE ein, der aus einem dreischichtigen GAT-Netzwerk besteht.
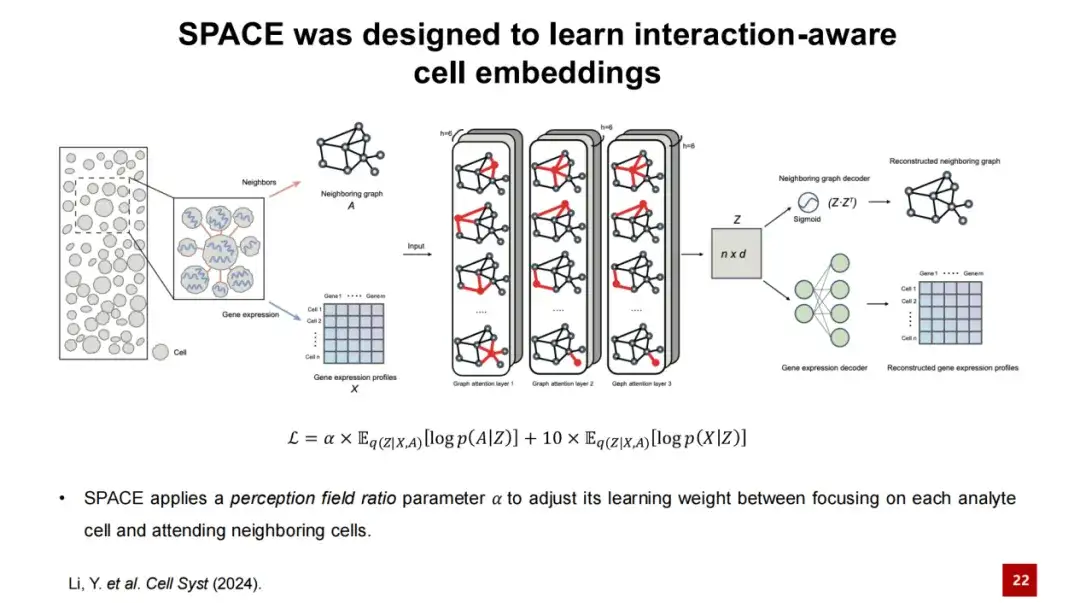
Durch die Verarbeitung des Encoders können wir die eingebettete Darstellung jedes Knotens erhalten und sie durch zwei unabhängige Decoder rekonstruieren:Ein Decoder rekonstruiert die niedrigdimensionale Darstellung der verborgenen Schicht der Zelle zurück in einen Nachbarschaftsgraphen, und der andere Decoder rekonstruiert das Genexpressionsprofil der Zelle. Die Verlustfunktion des SPACE-Modells ist die Summe dieser beiden Rekonstruktionsverluste.
In diesem ProzessWir entwerfen einen Wahrnehmungsfeldverhältnisparameter α, um die Gewichte der beiden Verlustfunktionen im Modell anzupassen.
Wenn der α-Wert klein ist,Das Modell konzentriert sich mehr auf die Rekonstruktion der Genexpression der Zelle selbst, und die erhaltene Zelleinbettung kann zur Identifizierung des Zelltyps verwendet werden.Wenn der α-Wert groß ist,Das Modell konzentriert sich stärker auf die Interaktion zwischen Zellen, und die zu diesem Zeitpunkt erhaltene Zelleinbettung kann zur Identifizierung von Gewebemodulen verwendet werden. Da die niedrigdimensionale Zelleneinbettung Z Informationen über Zellinteraktionen enthält, nennen wir die durch SPACE erhaltene niedrigdimensionale Einbettungsdarstellung „interaktionsbewusste Zelleneinbettungen“.
Um räumliche Zellsubtypen zu identifizieren, haben wir SPACE auf den Datensatz des primären Motorkortex von Mäusen angewendet.
In der folgenden Abbildung zeigt die obere linke Ecke die räumliche Position jeder Zelle im eigentlichen Gewebe, wobei ein Punkt eine Zelle darstellt und die Farbe den Zelltyp angibt. Dies ist eine UMAP-Karte, die auf der Grundlage der Genexpression generiert wurde. Die beiden Abbildungen unten links zeigen die von SPACE identifizierten räumlichen Zelluntertypen und ihre Positionen im Raum. Wir haben eine Konfusionsmatrixanalyse dieser räumlichen Zellsubtypen mit den in der ursprünglichen Studie bereitgestellten Zelltypen durchgeführt (siehe Abbildung rechts). Die Ergebnisse zeigten, dass die beiden im Allgemeinen übereinstimmten, mit einem angepassten Rand-Index (ARI) von 0,6. gleichzeitig, SPACE kann Astrozyten genauer von Oligodendrozyten unterscheiden und mehr Zellsubtypen identifizieren.
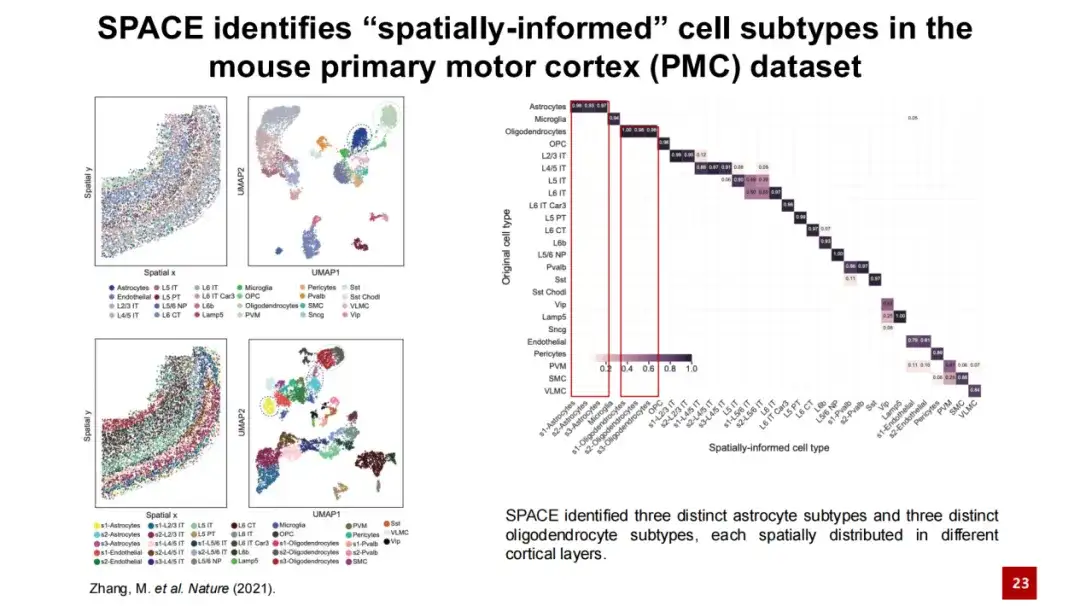
Das untere linke Bild zeigt die Organisationsstruktur des primären Motorkortex von Mäusen. Die Schicht stellt die kortikale Struktur dar und WM steht für weiße Substanz. Der geschichtete Aufbau von Schicht 1 bis zur weißen Substanz ist deutlich zu erkennen. Die drei von SPACE identifizierten Sternzellsubtypen waren allein anhand der Genexpression schwer zu unterscheiden und wurden in der UMAP-Karte vermischt.
Allerdings unterscheiden sich diese drei Zellsubtypen deutlich in ihrer räumlichen Verteilung: Der Zellsubtyp s1 ist hauptsächlich im Bereich von Schicht 1 bis Schicht 4 verbreitet, s2 ist hauptsächlich im Bereich von Schicht 5 bis Schicht 6 verbreitet und s3 ist hauptsächlich in der weißen Substanz verbreitet. Wir haben die Anteile der Zelltypen rund um diese drei Sternzellsubtypen gezählt und die Ergebnisse stimmten mit dieser Schichtungsregel überein. Obwohl die Genexpression der drei Zellsubtypen ähnlich war, zeigten sie dennoch ihre eigenen spezifischen Gene mit hoher Expression.

Die drei von SPACE identifizierten Sternzellsubtypen stimmen weitgehend mit früheren Studien überein. Frühere Studien haben gezeigt, dass es eine Interaktion zwischen Astrozyten und Neuronen gibt und dass die Schichtung der Astrozyten der Schichtung der Neuronen entspricht. Durch das Ausschalten von Schlüsselfaktoren in Neuronen stellten die Forscher fest, dass die Schichtstruktur der Neuronen zerstört wurde und sich auch die Schichtstruktur der Astrozyten entsprechend veränderte. Dies deutet darauf hin, dass zwischen Astrozyten und Neuronen eine räumlich spezifische Interaktion und eine räumlich spezifische Genregulation stattfindet.
Anhand dieses Beispiels können wir erkennen, dassSPACE kann räumliche Informationen effektiv nutzen und verschiedene biologische Zelltypen mit räumlichen Merkmalen genau identifizieren.
Im vorherigen Artikel wurde vorgestellt, dass SPACE die Optimierungsrichtung des Modells durch Anpassen des Wahrnehmungsfeldverhältnisparameters α ändert: Es kann den Eigenschaften der Zellen selbst mehr Aufmerksamkeit schenken, um Zelltypen zu identifizieren, oder den Interaktionsinformationen zwischen Zellen mehr Aufmerksamkeit schenken, um Gewebemodule zu entdecken.
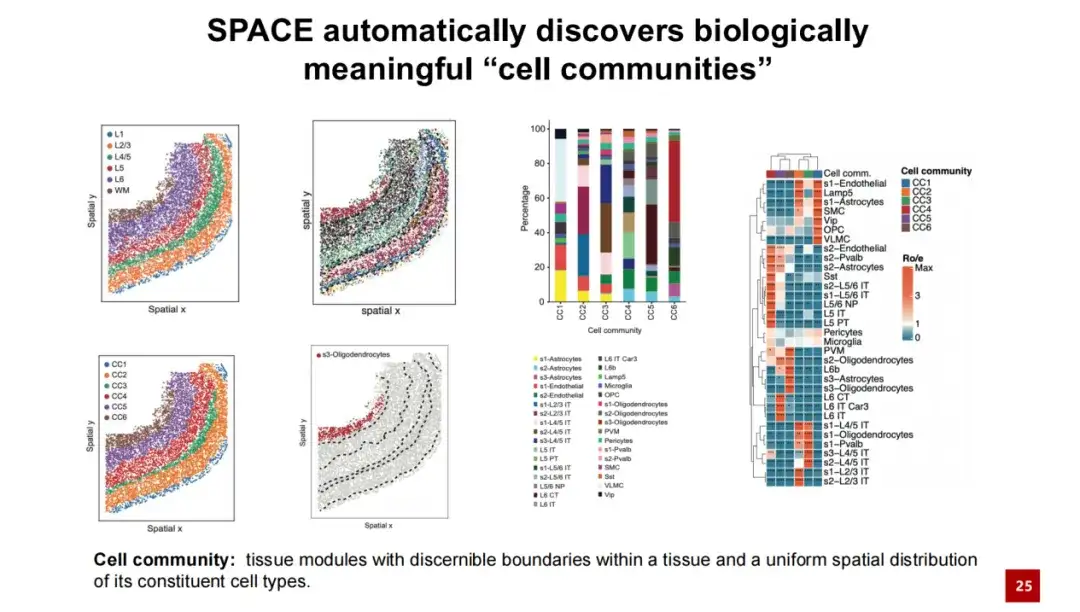
Im selben DatensatzDurch die Erhöhung des α-Wertes gelang es SPACE, das Gewebemodul zu entdecken.Wir nannten es Zellgemeinschaften (kurz CC). Wir glauben, dass die von SPACE entdeckten Gewebemodule erkennbare Grenzen haben und die räumliche Verteilung der Zelltypen in ihnen relativ gleichmäßig und konsistent ist. Wir verglichen die von SPACE entdeckten Zellgemeinschaften mit vorhandenen Gewebestrukturen und stellten fest, dass zwischen beiden eine gute Eins-zu-eins-Entsprechung besteht. Jede Zellkolonie enthält unterschiedliche Zelltypen und die räumliche Verteilung dieser Zelltypen innerhalb der Zellkolonie ist relativ gleichmäßig.
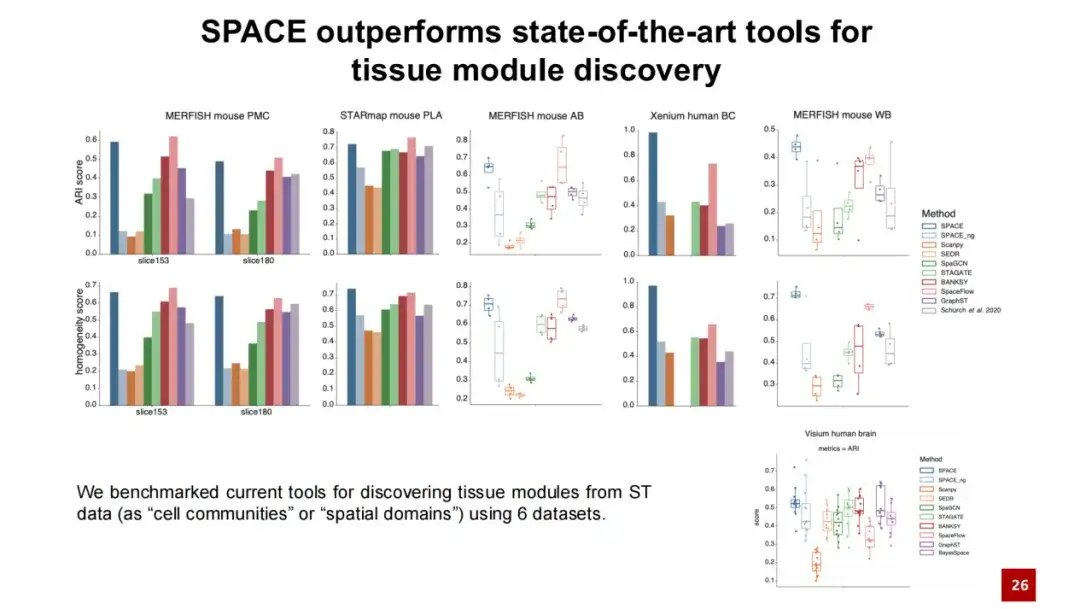
Wir haben SPACE mit bestehenden Methoden zur Entdeckung von Organisationsmodulen verglichen und Tests an fünf Datensätzen durchgeführt.Die Ergebnisse zeigen, dass SPACE in zwei Datensätzen die besten vorhandenen Methoden übertrifft und in den anderen drei Datensätzen eine mit den besten Methoden vergleichbare Leistung erbringt.Wir haben auch Tests und Analysen mit dem häufig verwendeten Visium-Datensatz zum menschlichen Gehirn durchgeführt und die Ergebnisse zeigten, dass SPACE auch auf räumliche Transkriptomdaten ohne Einzelzellauflösung anwendbar ist.
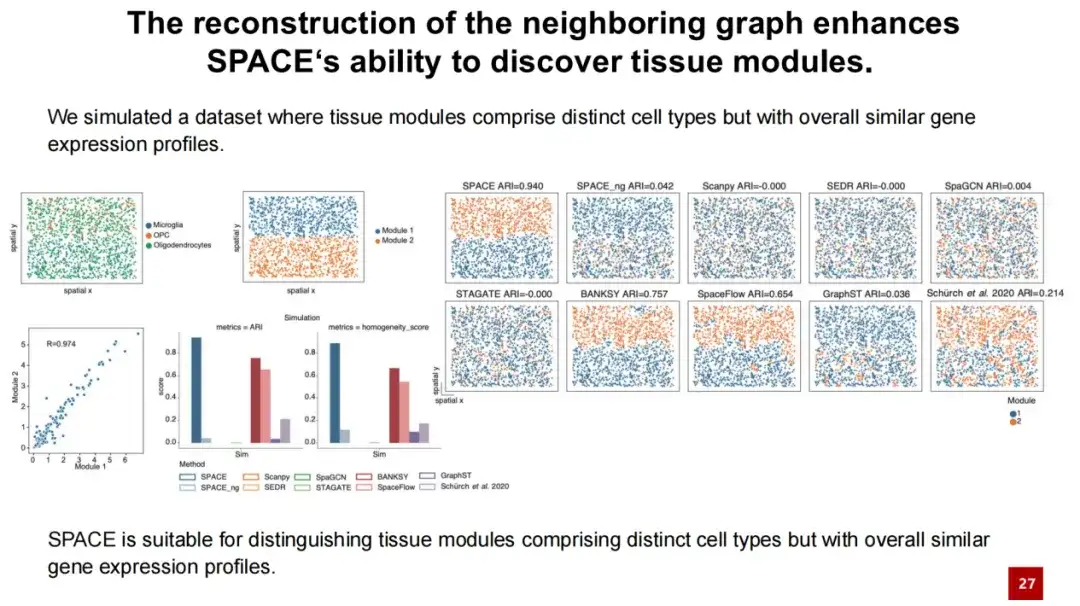
Darüber hinaus führen wir ein Testmodell namens SPACE_ng ein, wobei ng bedeutet, dass wir den Verlust der Rekonstruktion benachbarter Graphen im SPACE-Modell ausschalten. Die Ergebnisse zeigen, dass die Leistung von SPACE_ng der von SPACE weit unterlegen ist.
Um weiter zu veranschaulichen, dass SPACE die Leistung von Organisationsmodulen gut aus der Rekonstruktion des Nachbarschaftsdiagramms ermitteln kann, haben wir ein Simulationsexperiment entworfen. Wir haben Oligodendrozyten ausgewählt und Mikroglia und OPCs gleichmäßig zwischen den Oligodendrozyten verteilt (siehe Bild oben links unten), wodurch zwei Gewebemodule entstanden.
Da die meisten Zellen in diesen beiden Gewebemodulen Oligodendrozyten sind und eine sehr hohe Ähnlichkeit aufweisen (Kollaboration = 0,97), zeigen die Testergebnisse, dass SPACE anderen Methoden weit überlegen ist, während SPACE_ng nicht zwischen den beiden Gewebemodulen unterscheiden kann.Dies deutet darauf hin, dass die Leistung des SPACE-Gewebeerkennungsmoduls auf der Rekonstruktion des Nachbarschaftsgraphen beruht.
Wir haben in der nachfolgenden Analyse ein ähnliches Phänomen beobachtet, d. h., die Merkmale der durch SPACE identifizierten Zellgemeinschaften manifestieren sich nicht einfach als konsistente räumliche Genexpression wie in räumlichen Domänen, sondern spiegeln ähnliche Interaktionen zwischen benachbarten Zellen wider.
In der folgenden Heatmap stellt jede Spalte eine Zelle dar und ihre Farbe zeigt die Zellpopulation an, zu der die Zelle gehört, sowie ihren Zelltyp. Jede Zeile stellt einen Zelltyp dar und zeigt die relative Häufigkeit von Nachbarinteraktionen zwischen diesem Zelltyp und anderen Zellen. Aus dieser Heatmap können wir erkennen, dass Zellen, die zur gleichen Zellgemeinschaft gehören, Ähnlichkeiten in den Interaktionen mit Nachbarn aufweisen und dass diese Ähnlichkeit unabhängig vom spezifischen Zelltyp ist. Im Gegensatz dazu zeigten Zellen, die zu unterschiedlichen Zellpopulationen gehörten, größere Unterschiede in ihren Nachbarinteraktionen.
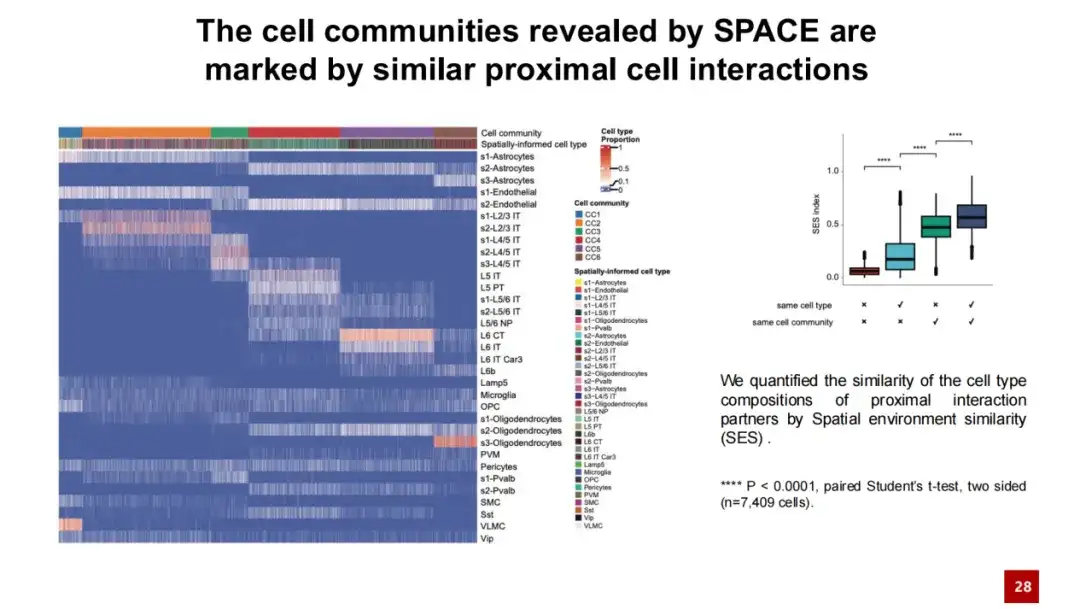
Darüber hinaus haben wir die Ähnlichkeit der Interaktionen zwischen Zellen mithilfe der Kosinus-Ähnlichkeit quantitativ berechnet. Die Ergebnisse zeigten, dass Zellen aus derselben Zellpopulation eine hohe Ähnlichkeit in ihren Interaktionen mit benachbarten Zellen aufwiesen, während Zellen aus unterschiedlichen Zellpopulationen relativ unterschiedliche extrazelluläre Interaktionen aufwiesen. Diese Ergebnisse zeigen, dassDie von SPACE entdeckten Zellgemeinschaften stellen nicht nur ein räumliches Genexpressionsmuster dar, sondern werden auch durch das proximale Zellinteraktionsnetzwerk beeinflusst.
Wir haben eine ähnliche Analyse in einem anderen Datensatz zur Mausplazenta durchgeführt. Das linke Bild zeigt die räumliche Position jedes Zelltyps im Datensatz, das mittlere linke Bild stellt die manuell annotierte Plazentagewebestruktur der Maus dar und das mittlere rechte Bild zeigt die fünf von SPACE entdeckten Zellpopulationen. Es ist ersichtlich, dass eine gute Eins-zu-eins-Entsprechung zwischen den von SPACE entdeckten Zellgemeinschaften und den manuell annotierten Gewebestrukturen besteht. Wir haben für jede Zellpopulation ein charakteristisches proximales Zellinteraktionsnetzwerk erstellt, wie in der Abbildung rechts dargestellt, das die einzigartigen Zell-Zell-Interaktionen innerhalb jeder Zellpopulation zeigt.

Am Beispiel von CC1 befindet sich die Gemeinschaft hauptsächlich im mütterlichen Deziduabereich. Wir fanden heraus, dass in CC1 eine starke Interaktion zwischen mütterlichen dekapsidierten S2-Zellen und S2-Glykotrophoblasten besteht. Frühere Studien haben gezeigt, dass während der Schwangerschaft von Mäusen Glykotrophoblasten in die mütterliche Dezidualregion eindringen und dort mit mütterlichen Dezidualzellen interagieren. Dadurch wird eine Umgestaltung der Arterien ausgelöst, die das mütterliche Blut zur Plazenta transportieren, ein Prozess, der für eine normale Schwangerschaft entscheidend ist.
Aus der obigen Analyse können wir schlussfolgern, dassSPACE kann Zell-Zell-Interaktionen in biologischen Proben identifizieren, die wichtige Auswirkungen auf Lebensprozesse haben.Daher spekulieren wir, dassDie von SPACE erstellten Interaktionsnetzwerke können zur Optimierung der Liganden-Rezeptor-basierten Zellkommunikationsanalyse verwendet werden.
Die Liganden-Rezeptor-basierte Zellkommunikationsanalyse ist eine gängige Methode in der Einzelzelldatenanalyse, d. h., basierend auf der Genexpression von Liganden und Rezeptoren zweier Zellen wird auf die Möglichkeit ihrer Zellkommunikation durch Liganden-Rezeptor-Paare geschlossen. Wir haben zunächst die Zell-Zell-Kommunikation in CC1 mithilfe von CellChat, einer häufig verwendeten Methode zur Analyse der Zellkommunikation, im Datensatz der Mausplazenta analysiert.
CellChat fand heraus, dass mütterliche dekapsidierte S3-Zellen über Signalwege wie Kollagen FN1 und THBS mit P-TGC-Zelltypen kommunizieren können. Allerdings ist für alle diese Signalwege ein physischer Kontakt erforderlich, damit es tatsächlich zustande kommt. Wir stellten jedoch fest, dass die beiden Zelltypen räumlich gesehen tatsächlich ziemlich weit voneinander entfernt sind (siehe untere rechte Ecke der Abbildung unten), sodass es unwahrscheinlich ist, dass sie tatsächlich in physischen Kontakt kommen.
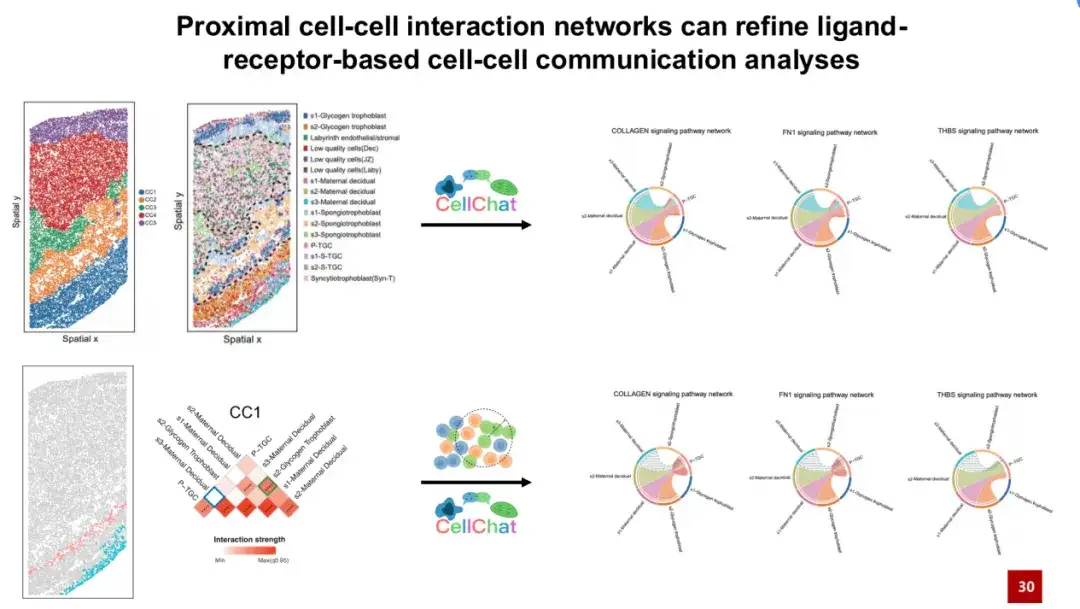
Dies wurde auch durch das in CC1 aufgebaute proximale Zellinteraktionsnetzwerk bestätigt. Die blauen Kästchen zeigen, dass Wechselwirkungen zwischen ihnen unwahrscheinlich sind.Durch die Einführung des von SPACE konstruierten charakteristischen proximalen Zellinteraktionsnetzwerks in die CellChat-Zellkommunikationsanalyse können wir Zellkommunikationssignale ausschließen, die im Weltraum eigentlich nicht auftreten können, und so falsch positive Signale wirksam reduzieren.
Karriere
Die Tsinghua-Universität und das State Key Laboratory of Membrane Biology haben in Hangzhou eine Zweigstelle für Membranstruktur und künstliche Intelligenzbiologie gegründet.. Derzeit rekrutiert das Team Fachkräfte, die sich mit interdisziplinärer Forschung zwischen künstlicher Intelligenz und Biologie beschäftigen. Wir laden Forscher, die sich für dieses Gebiet interessieren, herzlich ein, unserem Team beizutreten. Um weitere Einzelheiten zur Stellenbesetzung zu erhalten, scannen Sie bitte den untenstehenden QR-Code.









