Command Palette
Search for a command to run...
Ausgewählt Für Die ACL2024-Hauptkonferenz | InstructProtein: Angleichung Der Proteinsprache an Die Menschliche Sprache Durch Wissensanweisungen

Proteine sind die Grundlage für das Überleben von Zellen und kommen in allen Organismen vor, auch im menschlichen Körper. Es ist das Gerüst und die Hauptsubstanz, aus der Gewebe und Organe bestehen, und spielt eine zentrale Rolle bei chemischen Reaktionen, die für das Leben unerlässlich sind.
Angesichts der Komplexität und Variabilität der Proteinstruktur sind herkömmliche experimentelle Methoden zur Analyse der Proteinstruktur zeitaufwändig und mühsam. Es entstanden Proteinsprachmodelle (PLMs). Diese professionellen Modelle verwenden Aminosäuresequenzen als Eingabe und können Proteinfunktionen vorhersagen und sogar völlig neue Proteine entwerfen. Jedoch,PLMs sind zwar sehr gut darin, Aminosäuresequenzen zu verstehen, sie sind jedoch nicht in der Lage, die menschliche Sprache zu begreifen.
Auch große Sprachmodelle (LLMs) wie ChatGPT und Claude-2, die natürliche Sprache gut verarbeiten können, versagen, wenn es darum geht, die Funktion einer Proteinsequenz zu beschreiben oder Proteine mit spezifischen Eigenschaften zu erzeugen. Der Grund ist,Die aktuellen Proteintext-Datensätze weisen zwei große Mängel auf: Zum einen fehlen klare Anweisungssignale; das andere ist das Ungleichgewicht der Datenannotation. Zusammenfassend lässt sich sagen, dass in der aktuellen Forschung zu LLMs eine Lücke besteht, die nicht behoben wird: die Unfähigkeit, schnell zwischen menschlicher Sprache und Proteinsprache zu konvertieren.
Um diese Art von Problem zu lösen,Das von Huajun Chen und Qiang Zhang von der Zhejiang-Universität geleitete Team schlug das InstructProtein-Modell vor, das Wissensanweisungen verwendet, um die Proteinsprache an die menschliche Sprache anzupassen.Wir haben die wechselseitigen Generierungsfähigkeiten zwischen Proteinsprache und menschlicher Sprache untersucht, die Lücke zwischen den beiden Sprachen effektiv überbrückt und die Fähigkeit demonstriert, biologische Sequenzen in große Sprachmodelle zu integrieren.
Die Forschung trägt den Titel „InstructProtein: Angleichung der menschlichen und Proteinsprache durch Wissensvermittlung“.Akzeptiert von der ACL 2024-Hauptkonferenz.
Forschungshighlights:
* InstructProtein ist eine Studie zur Angleichung der menschlichen Sprache und der Proteinsprache durch Wissensanweisungen
* Erforschte die bidirektionalen Generierungsmöglichkeiten zwischen Proteinsprache und menschlicher Sprache und überbrückte so effektiv die Kluft zwischen den beiden Sprachen
* Experimente mit einer großen Anzahl bidirektionaler Protein-Text-Generierungsaufgaben zeigen, dass InstructProtein die Leistung bestehender hochmoderner LLMs übertrifft

Papieradresse:
https://arxiv.org/abs/2310.03269
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Umfassender wissenschaftlicher Datensatz
Das Korpus für die Vortrainingsphase des Modells enthält Proteinsequenzen von UniRef100 und Sätze aus PubMed-Abstracts.Basierend auf diesen Daten erstellten die Forscher einen Anweisungsdatensatz mit 2,8 Millionen Datenpunkten.
Während der Feinabstimmungsphase des Modells wurde der Protein-Wissensgraph mithilfe der von UniProt/Swiss-Prot bereitgestellten Anmerkungen erstellt, die Protein-Superfamilien, Familien, Domänen, konservierte Stellen, aktive Stellen, Bindungsstellen, Standorte, Funktionen und beteiligte biologische Prozesse umfassten; Die Daten für die wissenskausale Modellierung stammten aus den Datenbanken InterPro und Gene Ontology.
In der Modellbewertungsphase wählten die Forscher den Gene Ontology (GO)-Datensatz aus, um die Fähigkeit des Modells zur Annotation von Proteinfunktionen zu bewerten, und wählten dann Hu et al. aus. Datensatz zur Bewertung der Fähigkeit des Modells zur Vorhersage der Metallionenbindung (MIB).
Modellarchitektur: Feinabstimmung des vortrainierten Modells durch Erstellen eines Proteinwissens-Anweisungsdatensatzes
Um LLM die Fähigkeit zu verleihen, Proteinsprache zu verstehen, verwendet InstructProtein einen zweistufigen Trainingsansatz: zunächst ein Vortraining anhand von Protein- und natürlichen Sprachkorpora und dann eine Feinabstimmung mit einem etablierten Anweisungsdatensatz zum Proteinwissen.
Vortrainingsphase
Während der mehrsprachigen Vortrainingsphase wurden in dieser Studie große biologische Textdatenbanken verwendet, um das Sprachverständnis und den Wissenshintergrund des Modells im biologischen Bereich zu verbessern. Unter Mehrsprachigkeit versteht man die Fähigkeit, sowohl natürliche Sprachen (wie etwa englische Abstracts) als auch biologische Sequenzsprachen (wie etwa Proteinsequenzen) zu verarbeiten.
Phase der Modellfeinabstimmung
Während der Feinabstimmungsphase des ModellsDiese Studie schlägt eine Methode zur Datensatzkonstruktion namens „Wissensunterricht“ vor.Wissensgraphen (KGs) und große Sprachmodelle arbeiten zusammen, um einen ausgewogenen und vielfältigen Anweisungsdatensatz zu erstellen. Diese Methode ist nicht auf die Fähigkeit eines großen Sprachmodells angewiesen, die Proteinsprache zu verstehen. Dadurch werden durch Modellverzerrungen oder Halluzinationen verursachte Fehlinformationen vermieden. Der konkrete Bauprozess gliedert sich in drei Hauptphasen, wie in der folgenden Abbildung dargestellt:
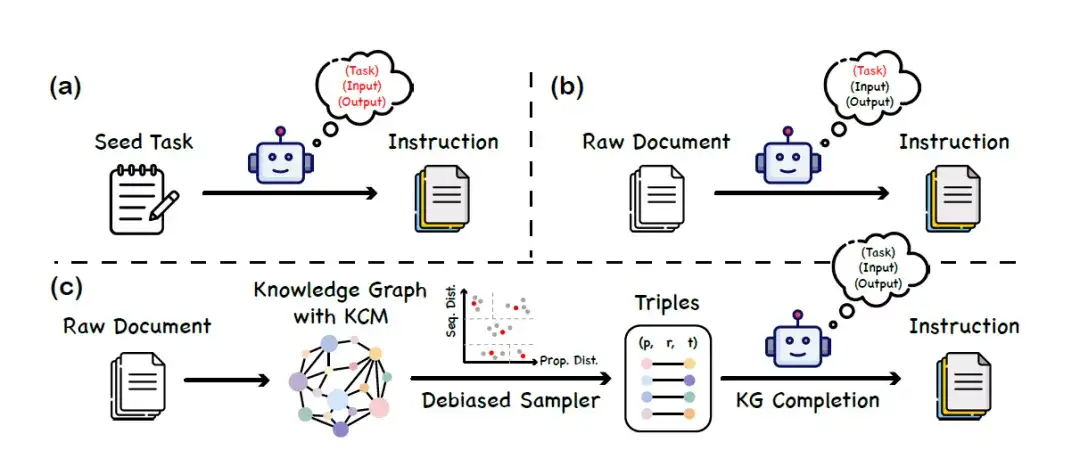
A. Fordern Sie LLM bei einer Reihe von Seed-Aufgaben auf, neue Anweisungsdaten zu generieren
B. Verwenden Sie LLM, um Anweisungsdaten zu generieren, die dem Inhalt des Rohdokuments entsprechen
C. Framework zur Anweisungsgenerierung basierend auf Wissensgraphen (KG)
* Aufbau des Wissensgraphen:Die Forscher nutzten UniProtKB als Datenquelle, um einen Protein-Wissensgraphen zu erstellen. Durch die Übernahme des Konzepts des Kettendenkens stellten die Forscher fest, dass es auch bei der Proteinannotation logische Ketten gibt. Beispielsweise hängen die biologischen Prozesse, an denen ein Protein beteiligt sein kann, eng mit seiner molekularen Funktion und subzellulären Lokalisierung zusammen, und die molekulare Funktion selbst wird von der Proteindomänenstruktur beeinflusst.
Um die Kausalkette dieses Proteinwissens darzustellen,Die Forscher führten ein neues Konzept namens Knowledge Causal Modeling (KCM) ein.Genauer gesagt besteht das kausale Wissensmodell aus mehreren miteinander verbundenen Tripel, die in einem gerichteten azyklischen Graphen organisiert sind, wobei die Richtung der Kante die kausale Beziehung darstellt. Das Diagramm ordnet Tripletts von der mikroskopischen Ebene (die Merkmale der Proteinsequenz wie etwa die Struktur abdeckt) bis zur makroskopischen Ebene (die die biologische Funktion abdeckt) an. Die folgende Abbildung zeigt den Prozess der Generierung sachlicher, logischer und vielfältiger Anweisungen anhand eines Triples mit KCM unter Verwendung eines großen Sprachmodells in Kombination mit einem Wissensgraphen zur Erledigung der Aufgabe.
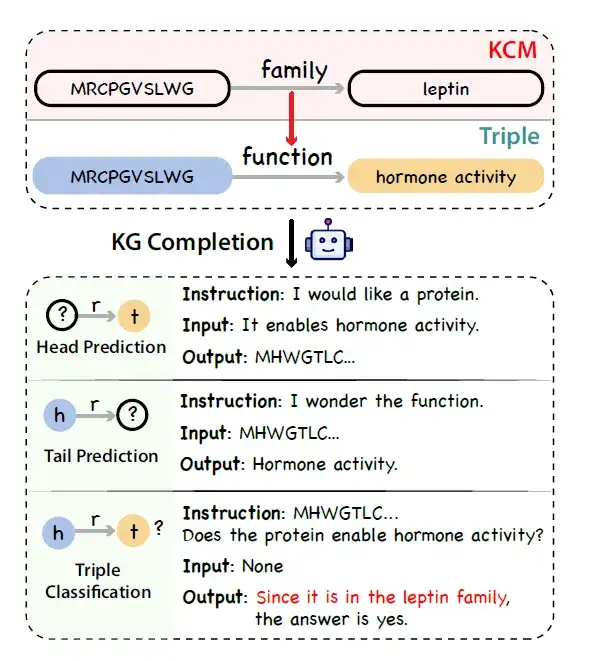
* Dreifachstichprobe des Wissensgraphen:Angesichts des Problems des Annotationsungleichgewichts in Wissensgraphen schlugen Forscher eine Strategie zur vorurteilsfreien Stichprobennahme als Alternative zur gleichmäßigen Stichprobennahme vor. Konkret werden Proteine zunächst nach ihrer Sequenz und Attributähnlichkeiten gruppiert und dann in jeder Gruppe gleichmäßig Tripletts extrahiert.
* Generierung von Anweisungsdaten:Die Forscher simulierten die Aufgabe der Wissensgraphenvervollständigung und verwendeten ein allgemeines LLM (wie ChatGPT), um Wissensgraphen-Tripel mit KCM in Anweisungsdaten umzuwandeln.
Dieser Ansatz ermöglicht die effiziente Erstellung eines umfangreichen und ausgewogenen Datensatzes mit Anweisungen zur Funktion und Lokalisierung von Proteinen, ohne sich zum Verständnis der Proteinsprache auf vordefinierte Modelle verlassen zu müssen.Bieten Sie zuverlässigere Datenunterstützung für die nachfolgende Erforschung und Anwendung von Proteinfunktionen.
Durch eine Kombination aus Vortraining und Feinabstimmung kann das resultierende Modell namens InstructProtein verschiedene Vorhersage- und Annotationsaufgaben im Zusammenhang mit Proteinsequenzen besser durchführen.Beispielsweise ist es möglich, die Funktion eines Proteins genau vorherzusagen oder es an einem bestimmten subzellulären Ort zu lokalisieren – was wichtige Auswirkungen auf die Proteintechnik, die Arzneimittelforschung und die biomedizinische Forschung im Allgemeinen hat.
Forschungsergebnisse: InstructProtein übertrifft bestehende hochmoderne LLMs
Die Studie bewertete umfassend die Fähigkeiten von InstructProtein im Bereich des Verständnisses und Designs von Proteinsequenzen:
Verständnis der Proteinsequenz
Die Forscher bewerteten die Leistung des InstructProtein-Modells anhand der folgenden drei Klassifizierungsaufgaben:Vorhersage der Proteinposition, Vorhersage der Proteinfunktion, Vorhersage der Fähigkeit zur Bindung von Proteinmetallionen. Diese Aufgaben ähneln Leseverständnisproblemen in natürlicher Sprache, wobei jeder Datensatz eine Proteinsequenz und eine Frage enthält und das Modell eine Ja/Nein-Frage beantworten muss. Alle Auswertungen werden im Zero-Shot-Setting durchgeführt.
Die Ergebnisse der Auswertung sind in der folgenden Tabelle dargestellt:InstructProtein erreicht bei allen Aufgaben im Vergleich zu allen Basismodellen eine hochmoderne Leistung.
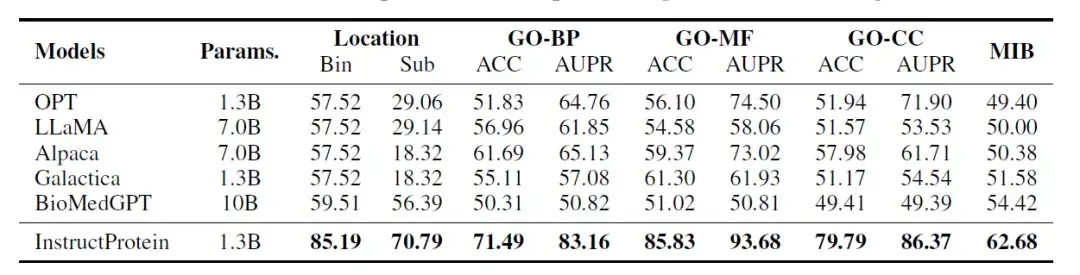
Darüber hinaus sind zwei wichtige Erkenntnisse bemerkenswert. Erstens übertrifft InstructProtein LLMs, die aus Trainingskorpora für natürliche Sprachen stammen (z. B. OPT, LLaMA, Alpaca), deutlich. Dies zeigt, dassDas Training mit einem Korpus, das sowohl Proteine als auch natürliche Sprache enthält, ist für LLMs von Vorteil und verbessert ihre Fähigkeiten im Verständnis von Proteinsprachen.
Zweitens: Obwohl sowohl Galactica als auch BioMedGPT UniProtKB als Korpus für die natürliche Sprache und Proteinausrichtung verwenden, übertrifft InstructProtein sie durchweg. Die Ergebnisse bestätigten, dassDie hochwertigen Unterrichtsdaten dieser Studie können die Leistung in Zero-Shot-Einstellungen verbessern.
Darüber hinaus waren LLMs (OPT, LLaMA, Alpaca und Galactica) bei der Aufgabe zur subzellulären Proteinlokalisierung (Bin) stark verzerrt, was dazu führte, dass alle Proteine in dieselbe Gruppe eingeordnet wurden, was zu einer Genauigkeit von 57,52% führte.
Proteinsequenzdesign
In Bezug auf das Proteindesign haben die Forscher eine Aufgabe zum „Instruction Protein Pairing“ entwickelt: Bei einem gegebenen Protein und seiner Beschreibung muss das Modell aus der entsprechenden Beschreibung und 9 nicht entsprechenden Beschreibungen das am besten geeignete auswählen.
Wie in der folgenden Tabelle gezeigt:InstructProtein übertrifft alle Basismodelle bei der Aufgabe der Befehls-Protein-Paarung deutlich.
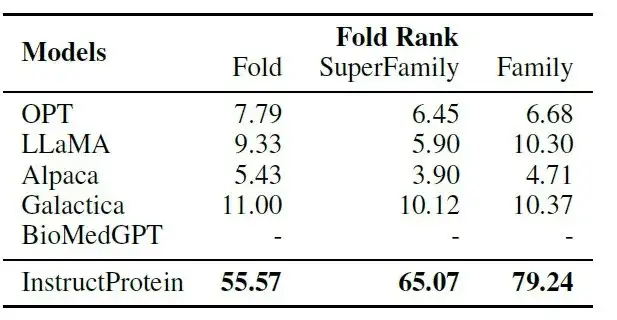
Unter diesen konzentriert sich BioMedGPT auf die Umwandlung von Proteinen in Text und verfügt nicht über Fähigkeiten zum Proteindesign. Galactica weist in der Zero-Shot-Einstellung der Ausrichtung von Anweisungen auf Proteine eine eingeschränkte Leistung auf, da es auf einem narrativen Proteinkorpus trainiert ist.Diese Ergebnisse bestätigen die Überlegenheit der Fähigkeiten des InstructProtein-Modells, Anweisungen bei der Proteinproduktion zu befolgen.
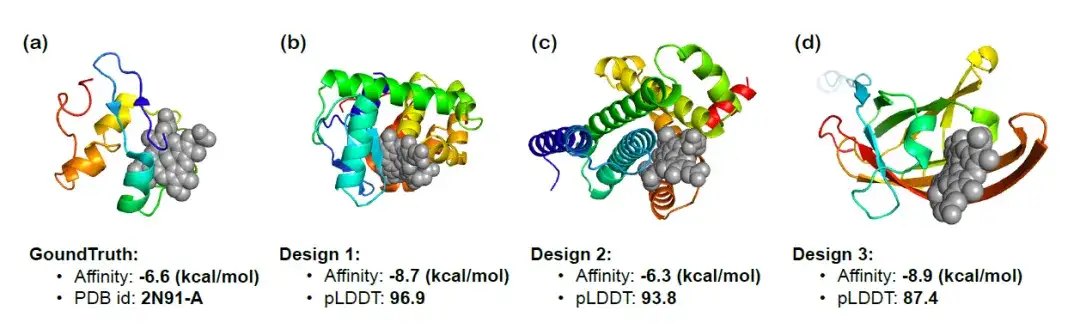
Um die Fähigkeit von InstructProtein, Proteine nach funktionell relevanten Anweisungen zu entwerfen, weiter zu validieren, verwendeten die Forscher InstructProtein, um Häm-bindende Proteine zu entwerfen, die an bestimmte Verbindungen binden konnten, und visualisierten die 3D-Strukturen der drei resultierenden Proteine. Die folgende Abbildung zeigt die Docking-Ergebnisse, die Vorhersage der Bindungsaffinität (je niedriger, desto besser) und den pLDDT-Score (je höher der absolute Wert, desto besser). Es wurde beobachtet, dass das resultierende Protein eine signifikante Bindungsaffinität aufwies,Die Wirksamkeit von InstructProtein beim Design von Häm-bindenden Proteinen wurde bestätigt.
Der Weg zur Erforschung von Proteinmodellen hat gerade erst begonnen
In den letzten Jahren haben große Sprachmodelle revolutionäre Veränderungen im Bereich der natürlichen Sprachverarbeitung bewirkt. Diese Modelle werden in vielen Bereichen des täglichen Lebens häufig verwendet, beispielsweise bei der Sprachübersetzung, der Informationsbeschaffung und der Codegenerierung. Obwohl diese Sprachmodelle bei der Verarbeitung natürlicher Sprache und kodierter Sprache gute Ergebnisse erzielen, sind sie mit biologischen Sequenzen (wie etwa Proteinsequenzen) nicht in der Lage.In diesem Zusammenhang kommt die Entstehung eines Protein-Large-Language-Modells gerade recht.
Das Protein-Big-Language-Modell wird speziell für proteinbezogene Daten trainiert, darunter Aminosäuresequenzen, Proteinfaltungsmuster und andere biologische Daten im Zusammenhang mit Proteinen. Daher sind sie in der Lage, die Struktur, Funktion und Wechselwirkungen von Proteinen genau vorherzusagen. Das Proteinsprachenmodell stellt die modernste Anwendung der KI-Technologie in der Biologie dar. Durch das Erlernen der Muster und Strukturen von Proteinsequenzen können die Funktion und Morphologie von Proteinen vorhergesagt werden, was für die Entwicklung neuer Medikamente, die Behandlung von Krankheiten und die biologische Grundlagenforschung von großer Bedeutung ist.
Im April 2023 zeigte eine in Science veröffentlichte Studie, dass Forscher des Meta-KI-Teams ein großes Sprachmodell verwendeten, das evolutionäre Informationen hervorbringen kann, um einen Sequenz-zu-Struktur-Prädiktor ESMFold zu entwickeln. Die Vorhersagegenauigkeit für Proteine mit Einzelsequenzen übertraf die von AlphaFold2, und die Vorhersagegenauigkeit für Proteine mit homologen Sequenzen lag nahe an der von AlphaFold2, und die Geschwindigkeit war um eine Größenordnung höher. Das Modell sagte mehr als 600 Millionen Metagenomproteine voraus und demonstrierte damit die Breite und Vielfalt natürlicher Proteine.
Im Juli 2023 schlugen Baidu Bio und die Tsinghua-Universität gemeinsam ein Modell namens xTrimo Protein General Language Model (xTrimoPGLM) mit einer Parameteranzahl von bis zu 100 Milliarden (100B) vor. In Bezug auf Verständnisaufgaben übertrifft xTrimoPGLM andere fortgeschrittene Basismodelle bei einer Vielzahl von Aufgaben zum Proteinverständnis deutlich. Im Hinblick auf Generierungsaufgaben ist xTrimoPGLM in der Lage, neue Proteinsequenzen zu generieren, die natürlichen Proteinstrukturen ähneln.

Link zum Artikel:
https://www.biorxiv.org/content/10.1101/2023.07.05.547496v3
Juli 2024Zhou Hao, ein assoziierter Forscher am Institute of Intelligent Industries der Tsinghua-Universität, arbeitete mit der Peking-Universität, der Nanjing-Universität und dem Shuimu Molecular Team zusammen, um ein mehrskaliges Proteinsprachenmodell ESM-AA (ESM All Atom) vorzuschlagen.Durch die Entwicklung von Trainingsmechanismen wie Residuenerweiterung und mehrskaliger Positionskodierung wurde die Fähigkeit zur Verarbeitung von Informationen im atomaren Maßstab erweitert. Die Leistung von ESM-AA bei Aufgaben wie der Ziel-Liganden-Bindung wurde deutlich verbessert und übertrifft aktuelle SOTA-Proteinsprachenmodelle wie ESM-2 und auch aktuelle SOTA-Lernmodelle für molekulare Repräsentationen wie Uni-Mol. Verwandte Forschungsergebnisse wurden auf der führenden Konferenz für maschinelles Lernen ICML unter dem Titel „ESM All-Atom: Multi-scale Protein Language Model for Unified Molecular Modeling“ veröffentlicht.

Papieradresse:
https://icml.cc/virtual/2024/poster/35119
Es muss betont werden, dass wir uns zwar in der Forschung zu großen Proteinsprachenmodellen erheblicher Fortschritte befinden, wir uns jedoch noch in der Anfangsphase der vollständigen Erfassung der Komplexität des Protein-Sequenzraums befinden. Beispielsweise steht das oben erwähnte InstructProtein-Modell vor Herausforderungen bei der Bewältigung numerischer Aufgaben, was insbesondere im Bereich der Proteinmodellierung wichtig ist, da dort quantitative Analysen erforderlich sind, darunter die Erstellung der 3D-Struktur, Stabilitätsbewertung und Funktionsbewertung. Zukunft,Die damit verbundene Forschung wird erweitert, um ein breiteres Spektrum an Anweisungen einschließlich quantitativer Beschreibungen einzubeziehen,Verbessern Sie die Fähigkeit des Modells, quantitative Ergebnisse zu liefern, und treiben Sie dadurch die Integration der Proteinsprache und der menschlichen Sprache voran und erweitern Sie seine Praktikabilität in verschiedenen Anwendungsszenarien.
Quellen:
1.https://arxiv.org/abs/2310.03269
2.https://mp.weixin.qq.com/s/UPsf9y9dcq_brLDYhIvz-w
3.https://hic.zju.edu.cn/ibct/2024/0228/c58187a2881806/page.htm








