Command Palette
Search for a command to run...
Dr. Zhou Bingxin Von Der Shanghai Jiao Tong University: Angesichts Der Herausforderung Knapper Biologischer Daten Gestalten Neuronale Graphennetze Das Verständnis Und Die Erzeugung Von Proteinen Neu

Am 12. August wurde die Sommerschule „AI for Bioengineering“ der Shanghai Jiao Tong University offiziell eröffnet und zog mehr als 100 Branchenkenner von über 30 Universitäten und 27 Unternehmen aus dem In- und Ausland an. Während des dreitägigen Studiums und Austauschs trafen sich zahlreiche Branchenexperten, Wirtschaftsvertreter und herausragende NachwuchswissenschaftlerEs wurde ein ausführlicher Austausch über die Integration und innovative Entwicklung von KI und Bioengineering angeboten.
Am Morgen des 12. sprach Zhou Bingxin, Assistenzforscher am Institut für Naturwissenschaften der Shanghai Jiao Tong University und am Shanghai National Center for Applied Mathematics (Zweigstelle der Shanghai Jiao Tong University), zum Thema „Vergangenheit und Gegenwart der künstlichen Intelligenz“. Es gliedert anschaulich die Entwicklungsgeschichte der KI und fasst die Merkmale des Meilensteinmodells zusammen.
Am Nachmittag sprach Dr. Zhou Bingxin im Gastexpertenbericht „Frontier Progress of Artificial Intelligence“ auch über das Thema „Graph Neural Networks and Protein Structure Representation“. Wir haben allen die Definition, Vorteile und neuesten Anwendungen von Graph-Neural-Networks in Bereichen wie der Proteinvorhersage und -generierung erläutert. HyperAI hat die Rede von Dr. Zhou Bingxin zu diesem Thema zusammengestellt und zusammengefasst, ohne die ursprüngliche Absicht zu verletzen. Nachfolgend finden Sie die Abschrift der Rede.
Nach Jahrzehnten rasanter Entwicklung hat Deep Learning verschiedene Modelle hervorgebracht, wie z. B. Convolutional Neural Networks, Recurrent Neural Networks und Transformer, mit denen Daten mit unterschiedlichen Eigenschaften verarbeitet werden können. Unter ihnen werden Graph-Neural-Netzwerke häufig in verschiedenen Szenarien verwendet, beispielsweise in sozialen Netzwerken, bei der Vorhersage von Flugbahnen und in der molekularen Modellierung usw., da sie Strukturdaten eingeben und verarbeiten können.
Viele Menschen sind jedoch der Meinung, dass Graph-Neural-Networks Graph-Convolutional-Networks (GCNs) sind, die keine komplexen Funktionen verarbeiten können und bei der Überlagerung mehrerer Schichten Probleme mit der übermäßigen Glättung haben können. Zudem unterliegen sie zahlreichen Einschränkungen. Da große Transformer-basierte Modelle zudem über starke Lernfähigkeiten bei großen Datensätzen verfügen,Warum also forschen und entwickeln wir weiterhin Graph-Neural-Networks?
Auf diese Fragen fasse ich die Antwort wie folgt zusammen: „Es ist SEXY.“
Das erste „S“ bedeutet, dass auf Graph-Neuronalen Netzwerken basierende Forschung gesund und nachhaltig ist. Wie in der folgenden Abbildung gezeigt, lässt sich durch den Vergleich des Kohlenstoffverbrauchs verschiedener menschlicher Verhaltensweisen erkennen, dass die Voraussetzung für die Leistungsfähigkeit des großen Modells ein enormer Energieverbrauch ist. Darüber hinaus wird durch die übermäßige Konzentration der Rechenressourcen und der Forschungsschwerpunkt auf große Modelle auch der Platz für andere Modellforschungen verengt. Auf lange Sicht werden nur große Unternehmen, die über ein Monopol auf Computerressourcen oder die Macht im öffentlichen Diskurs verfügen, in der Lage sein, Forschung und Entwicklung im Bereich der künstlichen Intelligenz aufrechtzuerhalten. Der wissenschaftliche Forschungsspielraum für Forscher in kleineren Unternehmen wird dagegen stark eingeschränkt sein.
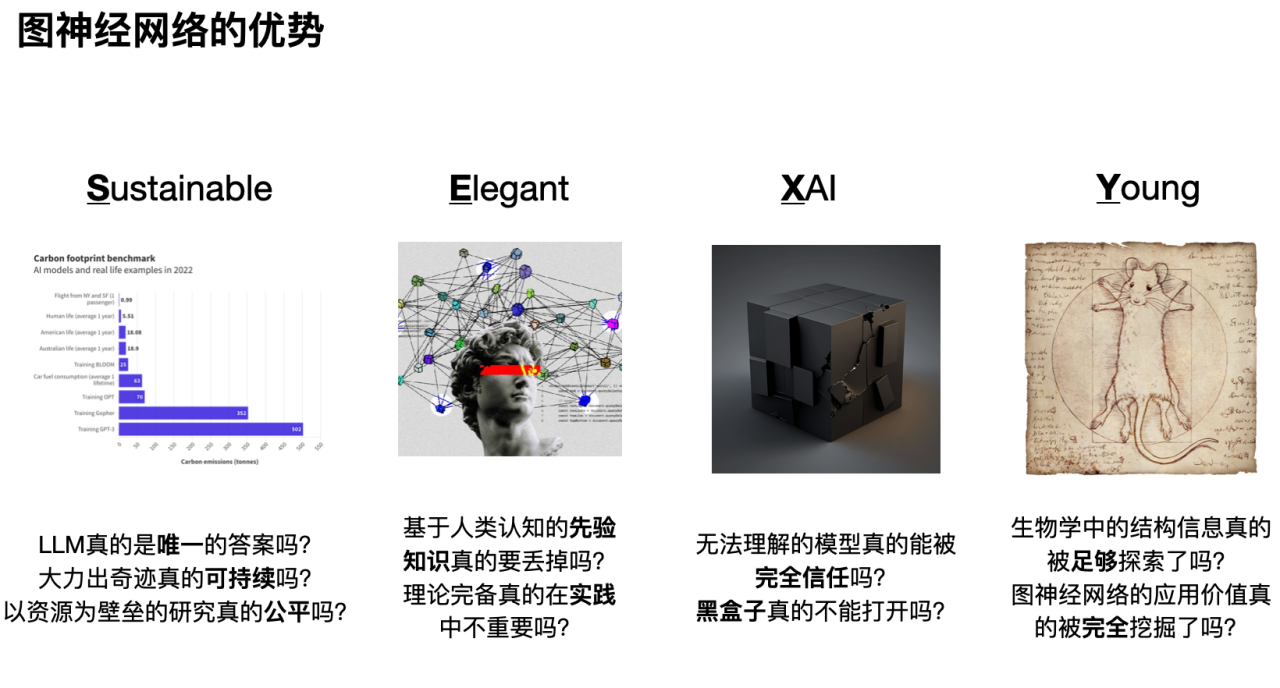
Das zweite „E“ bedeutet, dass die jahrhundertealten Erkenntnisse der Naturwissenschaften bei der rasanten Entwicklung der künstlichen Intelligenz nicht außer Acht gelassen werden dürfen. Zusätzlich zum Erlernen der Merkmalsdarstellung können Graph-Neuronale Netzwerke auch auf elegante Weise menschliches Vorwissen integrieren (induktive Verzerrung). Darüber hinaus verfügen Graph-Neural-Networks im Vergleich zu anderen datengesteuerten Modellen über mehr theoretische Unterstützung, beispielsweise in den Bereichen Signalverarbeitung, soziale Dynamik usw.
Das dritte „X“, Graph-Neural-Networks, trägt zur Verbesserung der Interpretierbarkeit von Deep-Learning-Netzwerken bei. Mit der Entwicklung künstlicher Intelligenz achtet man immer mehr auf die Aussagekraft und Rationalität von Modellergebnissen. Durch eingehende Forschung zur Interpretierbarkeit von Graph-Neural-Networks können wir die Logik und Grundlage hinter Modellentscheidungen besser verstehen und die Zuverlässigkeit und Vertrauenswürdigkeit des Modells verbessern.
Viertens, „Y“: Als junges und sich schnell entwickelndes Fachgebiet weisen Graph-Neuralnetzwerke noch immer eine große Anzahl ungelöster Probleme und Herausforderungen auf und bieten Forschern einen breiten Raum für Erkundungen. Darüber hinaus bieten Graph-Neuronale Netzwerke ebenso wie Convolutional-Neuronale Netzwerke für die Bildverarbeitung und Self-Attention-Mechanismen für die Verarbeitung natürlicher Sprache gute Lösungen für viele biologische Probleme (insbesondere für solche mit unzureichenden Daten und wichtigem Vorwissen).
Als Nächstes werde ich Ihnen den spezifischen Anwendungswert von Graph-Neural-Networks aus drei Blickwinkeln erläutern: molekulare Daten und Graph-Darstellung, Einführung in klassische Graph-Neural-Networks sowie Graph-Neural-Networks und weitere biologische Probleme.
Molekulare Daten und grafische Darstellung: Drei Elemente biologischer Datengrafiken
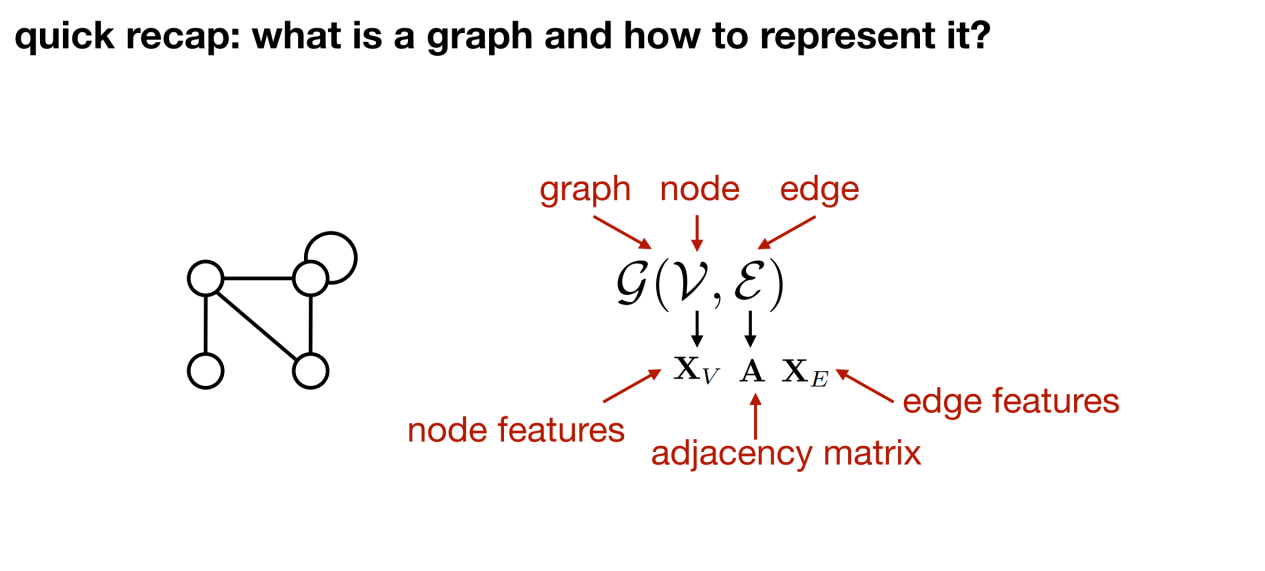
Wenn Sie biologische Daten in eine grafische Darstellung umwandeln möchten, müssen Sie zunächst die Frage beantworten: Was ist ein Graph und aus welchen Grundelementen besteht er? Allgemein gesprochen,Ein Graph enthält drei Elemente: Knoten; Kanten (Verbindungsbeziehungen zwischen Knoten); Graphen (die vollständige Einheit, die aus Knoten und Kanten besteht).
Wie verwenden wir diese drei Elemente, um das Studienobjekt in der Biologie zu definieren? Die folgende Abbildung zeigt 4 Fälle:

Für ein kleines Molekül (Abb. Jedes Atom kann als Knoten definiert werden und die Entfernungsbeziehung oder chemische Bindungsbeziehung zwischen Atomen kann durch Kanten dargestellt werden.
Wenn wir Protein auf der Aminosäureebene betrachten, Das Protein als Ganzes kann als Diagramm betrachtet werden, wobei jede Aminosäure einen Knoten im Diagramm darstellt. Wenn unterschiedliche Aminosäuren räumlich nahe beieinander liegen, kann man davon ausgehen, dass zwischen ihnen eine gewisse Korrelation besteht und diese räumlich nahen Aminosäureknoten durch Kanten verbunden sind.
Ähnlich verhält es sich mit der Sekundärstruktur von Proteinen. Dann kann jede Sekundärstruktur als Knoten im Proteingraphen betrachtet werden und ihre benachbarten oder räumlich nahen Sekundärstrukturen sind durch Kanten verbunden.
Und schließlich zum Wissensgraphen über Krankheiten: Verschiedene Krankheiten, Gene, Medikamente, Patienten und andere Elemente können als Knoten betrachtet werden, und die Verbindungen zwischen den Knoten stellen die komplexen Beziehungen zwischen ihnen dar, beispielsweise kann ein bestimmtes Medikament eine bestimmte Krankheit behandeln oder ein bestimmtes Gen verursacht eine bestimmte Krankheit.
Nachdem Sie einen Graphen definiert haben,Im nächsten Schritt muss überlegt werden, wie die Informationen im Diagramm beschrieben werden sollen, beispielsweise die Eigenschaften von Knoten und Kanten.
Wie in der folgenden Abbildung gezeigt, bestehen bestimmte Beziehungen zwischen den vier Knoten. Um diese Beziehungen genau zu beschreiben, kann eine Adjazenzmatrix A definiert werden. Bei der Verarbeitung verschiedener biologischer Daten kann die Adjazenzmatrix verwendet werden, um zu charakterisieren, ob kovalente Bindungen zwischen Atomen bestehen, oder um die Nachbarn der k-Ordnung einer bestimmten Aminosäure zu bestimmen.

Darüber hinaus kann jedem Knoten und jeder Kante eine Reihe von Attributen zugeordnet werden. Am Beispiel eines Aminosäureknotens können Knotenattribute charakteristische Informationen wie seinen Typ sowie seine physikalischen und chemischen Eigenschaften enthalten. Kanten dienen als Brücken, die Knoten verbinden, und können auch Merkmalsinformationen enthalten. Beispielsweise deckt der Merkmalsvektor an jeder Kante den Abstand zwischen zwei Aminosäuren (einschließlich Sequenzabstand und räumlicher Abstand) und die Grundlage für die Festlegung der Kante ab (basierend auf der räumlichen Struktur oder den atomaren chemischen Bindungen usw.). Diese Randfunktionen bieten eine detailliertere und tiefere Perspektive zum Verständnis der Beziehung zwischen Knoten.
Zusammenfassend:Jede strukturierte Einheit (wie beispielsweise ein Protein) kann als Graph dargestellt werden. Wie in der folgenden Abbildung gezeigt: G kann zur Darstellung des Graphen verwendet werden, v stellt den Knoten dar, ε stellt die Kante dar, Xv stellt die Merkmale auf dem Knoten dar, die Adjazenzmatrix A stellt die Knotenverbindung dar und Xe stellt die Merkmale der Kante dar.
Basierend auf den drei Grundelementen eines Graphen (Knoten, Kanten und Graphen) können die Vektordarstellungs- und Vorhersageaufgaben auf dem Graphen wie folgt klassifiziert werden:
- Vorhersage auf Knotenebene. Wenn Sie beispielsweise eine Protein-Sequenz entwerfen, sagen Sie anhand eines bekannten Proteindiagramms den Aminosäuretyp voraus, der durch jeden Knoten im Diagramm dargestellt wird.
- Linkvorhersage. Bestimmen Sie anhand eines Graphen und aller Knoten, ob Beziehungen zwischen Knoten bestehen, wie etwa Genregulationsnetzwerke, Arzneimittelwissensgraphen und andere Vorhersageaufgaben.
- Graphvorhersage (Vorhersage auf Graphebene). Wenn sowohl Knoten als auch Kanten bestimmt sind, werden mehrere Graphen gleichzeitig gelernt und analysiert, um die Beschriftungen jedes Graphen vorherzusagen.
Was sind Graph Neural Networks: Nicht nur GCN, sondern auch GAT, GraphSAGE, EGNN und mehr
Graph-Neuralnetze suchen basierend auf den Verbindungsbeziehungen zwischen gegebenen Knoten nach Darstellungen der verborgenen Schichten jedes Knotens und finden für jeden Knoten eine Vektordarstellung. Im Vergleich zu anderen Datentypen besteht das wichtigste Merkmal eines Diagramms darin, dass es klar anzeigen kann, welche Knoten direkt miteinander verbunden sind und wie eng die Beziehungen zwischen den verschiedenen Knoten sind.Daher liegt die Essenz von Graph-Neuralnetzwerken in der Verwendung dieser induktiven Vorurteile und der Übermittlung von Nachrichten zwischen verbundenen Knoten. Je näher die Nachbarknoten beieinander liegen, desto größer ist ihr Einfluss auf den Zentralknoten.
Als Nächstes werde ich Ihnen mehrere klassische Graph-Convolutional-Neural-Networks vorstellen.
Das erste ist das Graph Convolutional Neural Network GCN, Wie in der folgenden Abbildung dargestellt, besteht der Kern darin, dass jede GCN-Schicht die Informationen der Nachbarn erster Ordnung zum zentralen Knoten durchschnittlich aggregiert und die aggregierten Informationen als neue Darstellung des zentralen Knotens verwendet.
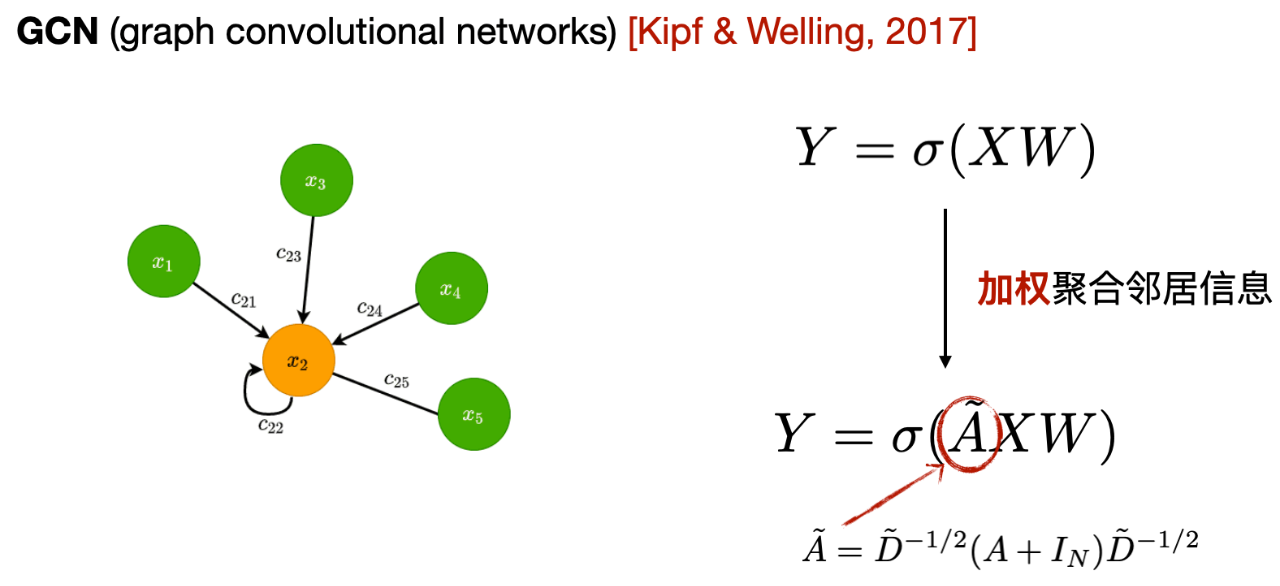
Aus dem Ausdruck können wir erkennen, dass der Unterschied zwischen GCN und MLP darin besteht, dass GCN die Adjazenzmatrix hinzufügt und die Nachbarinformationen erster Ordnung verwendet, um die Knotendarstellung zu aktualisieren. Darüber hinaus fügt es Selbstschleifen hinzu, um seine eigenen Informationen beim Aggregieren von Informationen zu stärken, und führt eine gewichtete Mittelwertbildung basierend auf der Anzahl der Nachbarn jedes Nachbarknotens durch.
- Nachbarn erster Ordnung: Der zentrale Knoten ist direkt mit anderen Knoten verbunden, d. h. die Punkte, die über eine Kante erreicht werden können, werden als Nachbarn erster Ordnung bezeichnet.
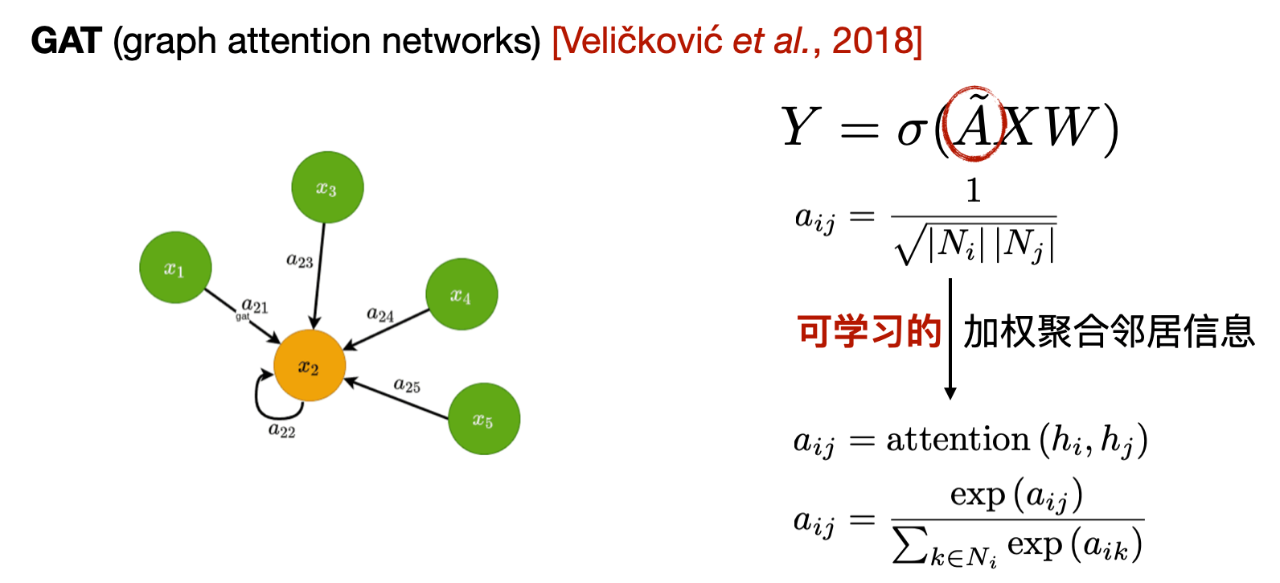
Das zweite ist das Graph Attention Network (GAT). Im Vergleich zu GCN besteht die Hauptänderung von GAT in der Art und Weise der Gewichtungsberechnung beim Aggregieren von Nachbarinformationen. GCN verwendet Gewichte, die auf Grundlage der Adjazenzmatrix berechnet werden, während GAT ein lernbares Gewicht auf Grundlage der Eigenschaften benachbarter Knoten berechnet.
Die beiden oben genannten Methoden sind typische Vertreter transduktiver Methoden. Sie erfordern einen vollständigen Graphen als Eingabe, was die Rechenkomplexität erhöht.In dieser Hinsicht schlägt GraphSAGE einen induktiven Ansatz vor. Bei jeder Informationsübertragung müssen nur die Nachbarn erster Ordnung des zentralen Knotens verstanden werden, und nur ein Teil der Nachbarinformationen wird zufällig zur Aggregation ausgewählt.
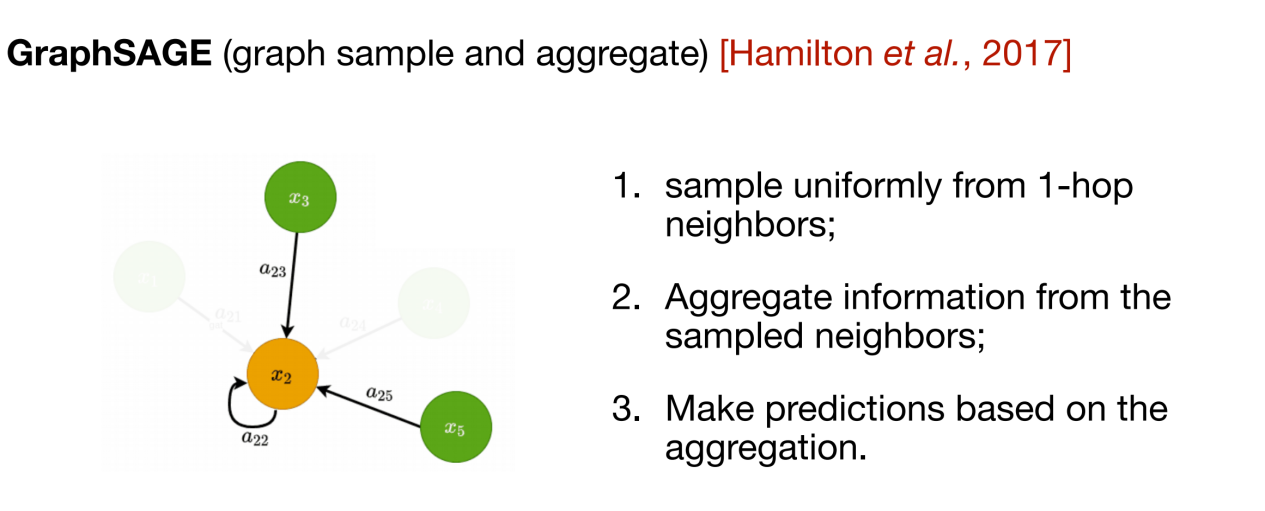
Die oben genannten drei Methoden dienen dazu, die Darstellung der Knoten im zweidimensionalen topologischen Strukturdiagramm zu aktualisieren, und das nachfolgende Nachrichtenübermittlungsnetzwerk (MPNN) integriert diese Art von Methode zur Informationsaggregation in ein Framework. Bei vielen biologischen Daten (wie etwa Molekülen) muss jedoch auch die dreidimensionale Struktur berücksichtigt werden.Zur Integration räumlicher Informationen können äquivariante Graph-Neural-Networks (EGNN) verwendet werden. Wie in der folgenden Abbildung dargestellt, besteht der Kern dieser Methode darin, dass zusätzlich zu den Merkmalsinformationen des Knotens selbst auch die relative Positionsbeziehung zwischen Knoten eingeführt wird, um die Rotationsäquivarianz und Translationsinvarianz der gelernten Darstellung sicherzustellen.

Darüber hinaus gibt es viele fortschrittliche Designs für neuronale Graphnetzwerke. Einige Designs können nicht nur die Vorhersageleistung des Modells verbessern, sondern konzentrieren sich auch auf die Verbesserung der Effizienz, die Reduzierung der Überglättung, das Hinzufügen einer mehrskaligen Darstellung und andere Anforderungen. Durch die Einführung kontinuierlicher Nachrichtenübermittlung, spektraler Graph-Faltungsmethoden usw. können auch ausdrucksstärkere Graph-Neuralnetzwerke für bestimmte Probleme bereitgestellt werden.
Wichtige Anwendungen von Graph-Neuronalen Netzwerken: am Beispiel der Vorhersage von Proteineigenschaften und der Sequenzgenerierung
Als Nächstes werde ich Ihnen die Anwendung von Graph-Neural-Networks beim Lernen von Proteinrepräsentationen erläutern.Hier teile ich es in zwei Kategorien ein: Vorhersagemodelle und Generierungsmodelle.
Kodierung und Vorhersage von Proteinmerkmalen
In Bezug auf Vorhersageaufgaben berücksichtigen wir drei Arten von Aufgaben: Vorhersage von Mutanteneigenschaften, Vorhersage der Löslichkeit und Subgraph-Matching, also vier spezifische Aufgaben.
Die erste Arbeit befasst sich mit der Vorhersage von Mutationsaufgaben. Wie in der folgenden Abbildung gezeigt, haben wir äquivariante Graph-Neuralnetzwerke verwendet, um die internen räumlichen Beziehungen von Proteinaminosäuren zu charakterisieren, wobei jeder Knoten eine Aminosäure darstellt und den Typ, die physikalischen und chemischen Eigenschaften sowie andere Merkmale der Aminosäure an diesem Punkt angibt. Die Kantenverbindungen im Diagramm spiegeln die Beziehungen zwischen Aminosäuren wider, beispielsweise das Potenzial für eine gemeinsame Evolution und den Einfluss gegenseitiger Kräfte.
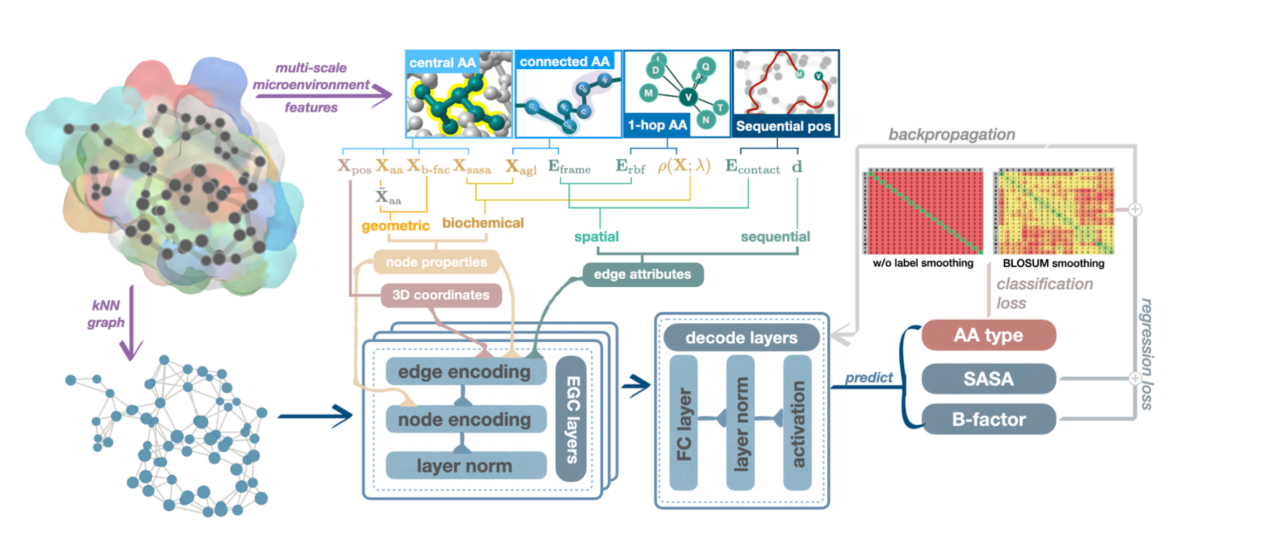
Anschließend verwendeten wir ein Vorhersagemodell, um verschiedene Mutanten zu bewerten und Mutationskombinationen mit hoher Punktzahl zu identifizieren, die die Proteineigenschaften am wahrscheinlichsten optimieren. Dieses leichtgewichtige Graph-Neuralnetzwerk kann die Trainings- und Datenkosten erheblich senken, indem es Aminosäuren und die Beziehungen zwischen Aminosäuren integriert, wodurch das Modell klein und schön wird und gleichzeitig eine hohe Leistung beibehält. Darüber hinaus hat die Überprüfung verschiedener Proteineigenschaften durch Nassexperimente gezeigt, dass dieses Modell die Wirkung und Erfolgsrate der gerichteten Evolution deutlich verbessern kann. Die Forschung trug den Titel „Protein Engineering with Lightweight Graph Denoising Neural Networks“ und wurde in ACS JCIM veröffentlicht.
Papieradresse:
https://pubs.acs.org/doi/10.1021/acs.jcim.4c00036
Die zweite Aufgabe besteht darin, auf der Grundlage der Strukturcodierung eine weitere Proteinsequenzcodierung hinzuzufügen. Dies liegt daran, dass strukturelle Informationen davon ausgehen, dass die Wechselwirkungen zwischen benachbarten Aminosäuren stärker sind, während die Wechselwirkungen über große Entfernungen hinweg extrem schwach sind. Diese Annahme trifft nicht ganz auf die tatsächliche Situation zu, daher sind Sequenzinformationen erforderlich, um die Betrachtung von Wechselwirkungen über große Entfernungen zu ergänzen. Darüber hinaus werden bei Informationen zu unterschiedlichen biologischen Eigenschaften unterschiedliche Schwerpunkte gesetzt. Für die Bindungsenergie und die thermische Stabilität spielen Strukturinformationen eine dominierende Rolle, wenn es jedoch um Eigenschaften wie die katalytische Aktivität geht, sind Informationen über die Art der Aminosäuren entscheidender.
Wie in der Abbildung unten gezeigt, haben wir experimentelle Tests mit mehr als 200 Assays auf ProteinGym durchgeführt und die beste Leistung der Nicht-MSA-Methoden erzielt. Die Studie wurde in eLife unter dem Titel „Semantical and Geometrical Protein Encoding Toward Enhanced Bioactivity and Thermostability“ veröffentlicht.
Papieradresse:
https://elifesciences.org/reviewed-preprints/98033
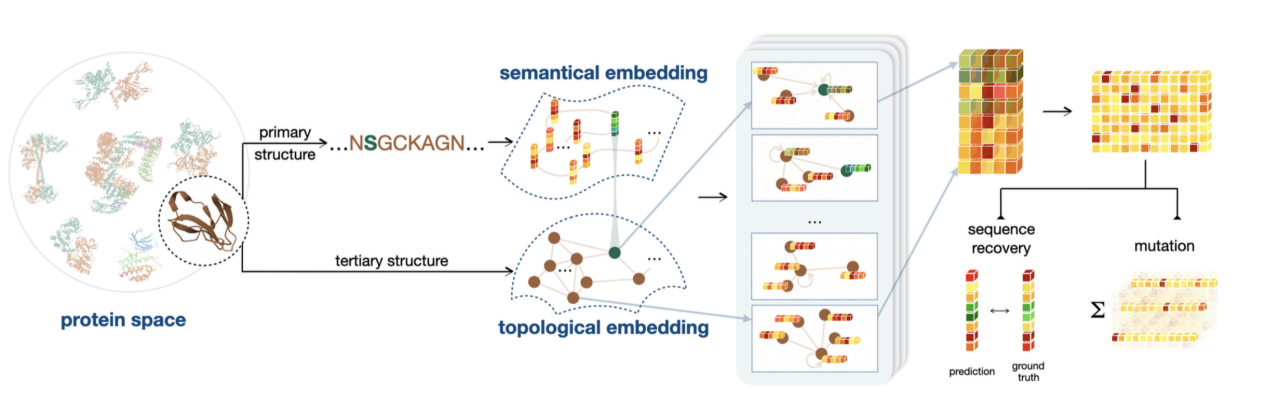
Das Codierungsmodul auf Aminosäureebene der dritten Arbeit stimmt mit dem der zweiten Arbeit überein. Die Informationsintegration basiert auf der Sequenz und Struktur von Proteinen. Der Unterschied besteht darin, dass auch verschiedene Informationen auf Proteinebene integriert werden, die auf Vorkenntnissen basieren, wie z. B. Proteinlänge, proportionale Verteilung von 20 Aminosäuren usw.
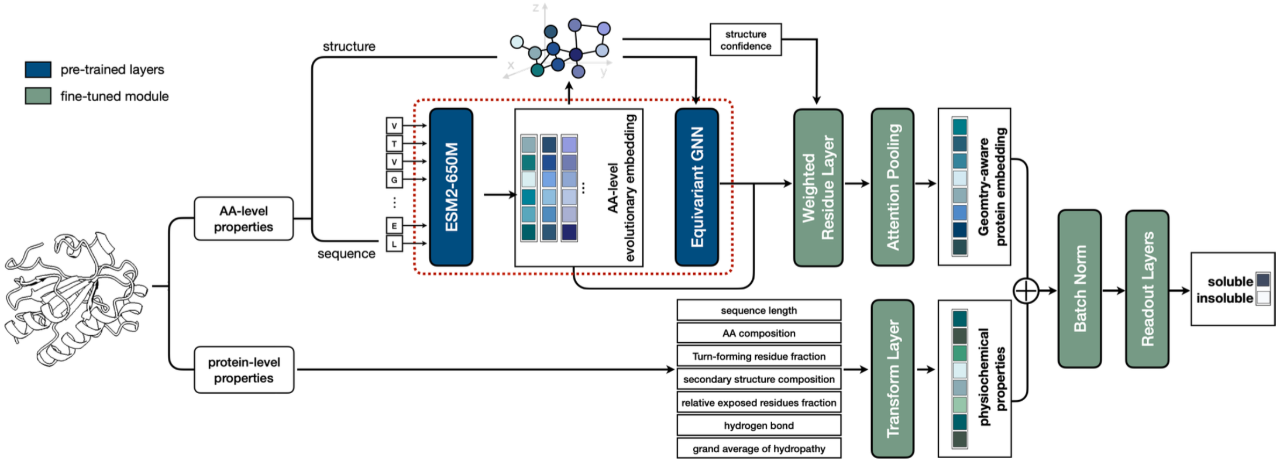
Wie in der folgenden Abbildung dargestellt, haben wir die Vorhersagewirkung des Modells auf die Proteinlöslichkeit getestet und auf Grundlage von Berechnungen und Experimenten SOTA-Ergebnisse anhand von Tausenden von Testdaten erzielt. Die Forschung mit dem Titel „ProtSolM: Protein Solubility Prediction with Multi-modal Features“ wurde von IEEE BIBM2024 (CCF Category B Conference) angenommen.
Adresse des Vorabdrucks:
https://www.arxiv.org/abs/2406.19744
Die vierte Aufgabe besteht darin, die lokalen Ähnlichkeiten von Proteinstrukturen zu erforschen. Wie in der Abbildung unten gezeigt, liegt der Kern des Proteins möglicherweise in bestimmten lokalen Strukturmerkmalen, auch wenn es insgesamt groß ist. Darüber hinaus können zwei Proteine aus makroskopischer Sicht zwar eine völlig unterschiedliche Sequenz und Struktur aufweisen, aber über ähnliche oder sogar identische Kernfunktionsmodule verfügen.
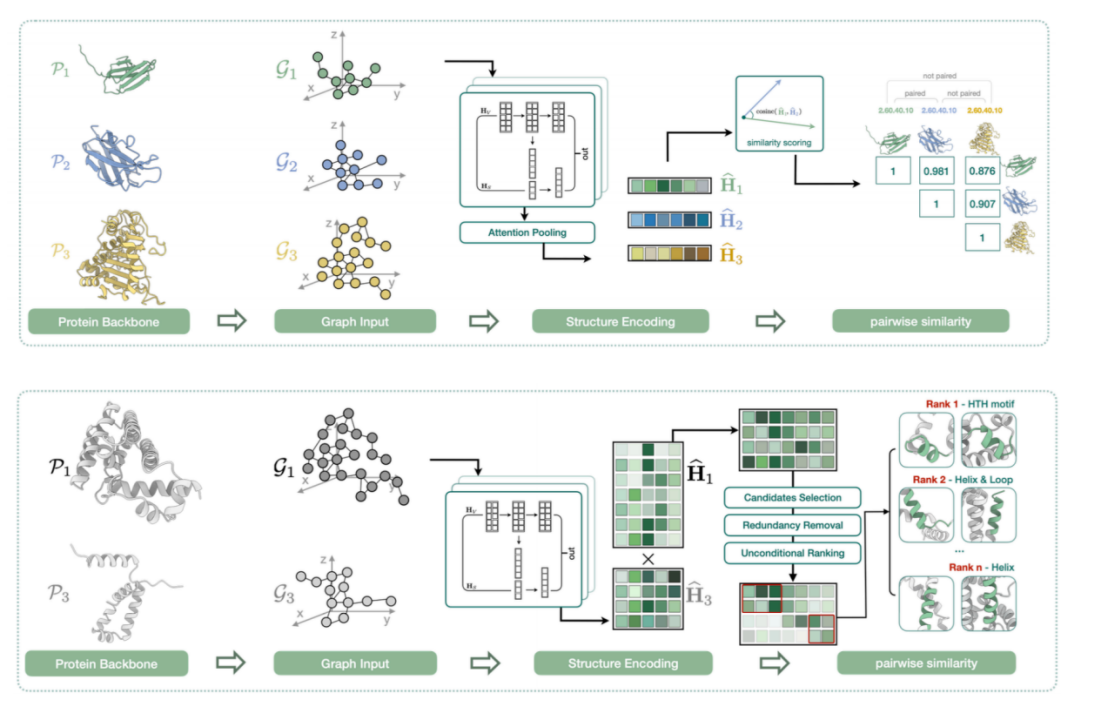
Daher finden wir eine implizite Darstellung der lokalen Struktur jedes Proteins und berechnen die Ähnlichkeit zwischen diesen Vektoren. Zusätzlich zum Vergleich der Eins-zu-eins-Ähnlichkeit zwischen Strukturen bewerten wir auch, ob es zwischen zwei vollständigen Proteinen ausrichtbare lokale Strukturfragmente gibt. Die Forschung mit dem Titel „Protein Representation Learning with Sequence Information Embedding: Does it Always Lead to a Better Performance?“ wurde von IEEE BIBM2024 angenommen.
Adresse des Vorabdrucks:
https://arxiv.org/abs/2406.19755
Sequenzgenerierung
Als nächstes werde ich zwei Arbeiten mit Ihnen teilen, nämlich das Entwerfen geeigneter Aminosäuresequenzen für Proteinstrukturen. Die Kernmodelle dieser beiden Arbeiten sind Diffusionswahrscheinlichkeitsmodelle (Diffusion).
Die erste Aufgabe besteht darin, auf Grundlage des bekannten Aminosäuregerüsts eine vollständige Proteinsequenz zu entwerfen, um die Proteinleistung zu verbessern. Der Modellrahmen ist in der folgenden Abbildung dargestellt. Im Gegensatz zur gerichteten Evolution haben wir Hunderte von Aminosäuren gleichzeitig modifiziert und Proteinsequenzen mit höherer Diversität erhalten. Einerseits könnte diese Methode einen völlig neuen Ausgangspunkt für die Evolution finden und Probleme wie lokale Optimalität und negative Aufwärtseffekte vermeiden, die bei der gerichteten Evolution häufig auftreten. Andererseits wird es durch die Modifizierung weiterer Aminosäuren möglich, Proteine mit geringerer Sequenzähnlichkeit, aber gleicher Funktion zu erhalten und so die Patentblockade zu durchbrechen.
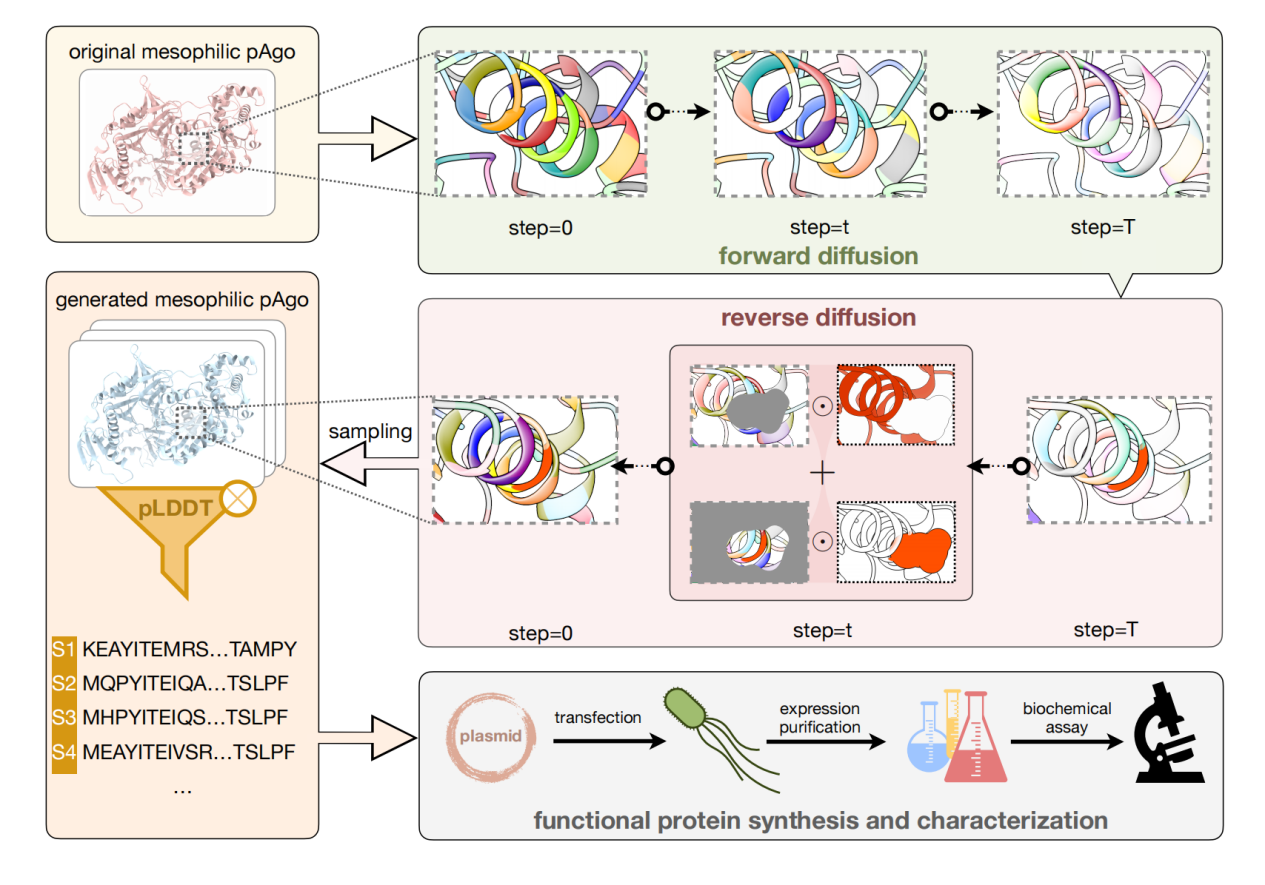
Wir verwendeten zwei Argonaute-Proteine (die bei mittleren bzw. ultrahohen Temperaturen arbeiten) als Designvorlagen und die meisten der über 40 generierten Proteine können DNA-Spaltung bei Raumtemperatur durchführen. Das beste Design weist eine mehr als zehnmal höhere Spaltaktivität als der Wildtyp auf und auch seine thermische Stabilität ist deutlich verbessert. Die Studie mit dem Titel „Conditional Protein Denoising Diffusion Generates Programmable Endonucleases“ wurde in Cell Discovery veröffentlicht.
Adresse des Vorabdrucks:
https://www.biorxiv.org/content/10.1101/2023.08.10.552783v1
Die zweite Aufgabe besteht, wie in der folgenden Abbildung dargestellt, darin, die Anzahl und Position der einzufüllenden Aminosäuren basierend auf der Sekundärstruktur unabhängig zu bestimmen, ohne die Struktur des Aminosäurerückgrats streng einzuschränken. Verglichen mit der auf dem Skelett basierenden Generierungsmethode kann durch diese gröbere Generierungsbedingung eine Sequenzdiversität in die generierte Sequenz eingeführt werden und sie kann auch die spezifischen Anforderungen der Proteinmodifikation und des neuen Designs erfüllen (beispielsweise ist bei Transmembranproteinen nur der Transmembranteil auf eine Helixstruktur beschränkt, die Länge und das spezifische Skelett dieses Teils sind jedoch nicht streng eingeschränkt). Die Studie mit dem Titel „Secondary Structure-Guided Novel Protein Sequence Generation with Latent Graph Diffusion“ wurde von ICML AI4Science angenommen und der vollständige Text wird derzeit geprüft.
Adresse des Vorabdrucks:
https://arxiv.org/html/2407.07443v1
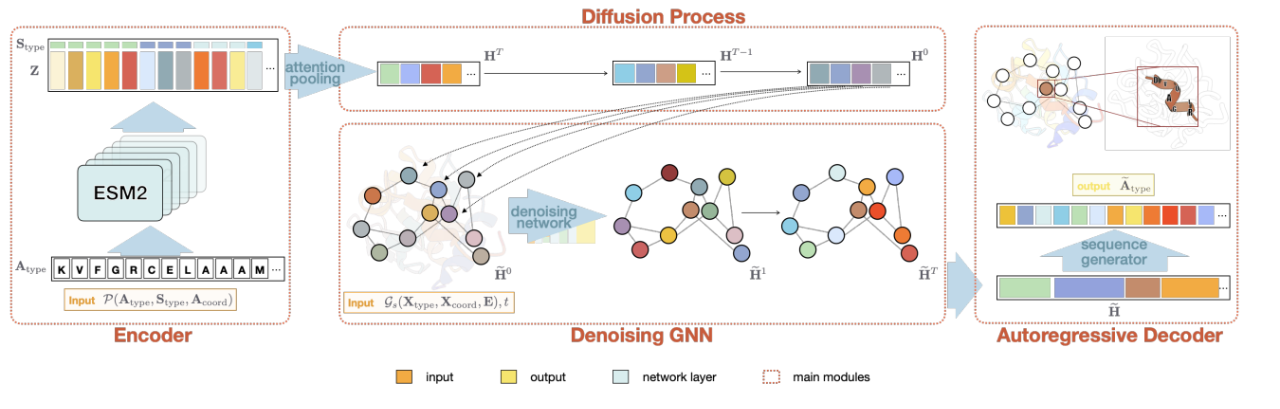
Die beiden oben genannten Arbeiten zum Design von Protein-Sequenzen auf Grundlage von Diffusion können entweder eine ganze Sequenz entsprechend dem Proteinskelett erzeugen oder einige Schlüsselaminosäuren und die Skelettstruktur fixieren und sie als Erzeugungsbedingungen verwenden, um die Aminosäuresequenz des nicht fixierten Teils auszufüllen.
Anwendung von Graph Neural Networks auf weitere biologische Probleme
Zusätzlich zur herkömmlichen molekularen Graphenmodellierung können Graph-Neural-Networks auch auf andere Daten- und Problemtypen angewendet werden, um die Erforschung biologischerer Probleme zu fördern. Als nächstes werde ich zwei Beispiele nennen.
Das erste Beispiel ist die Analyse und Vereinfachung biologischer sozialer Netzwerke. Ähnlich wie die komplexen Beziehungen in menschlichen sozialen Netzwerken gibt es in biologischen sozialen Netzwerken auf verschiedenen Ebenen (wie mikrobiellen Netzwerken, Gennetzwerken usw.) viele Inhalte, die es wert sind, erforscht zu werden.
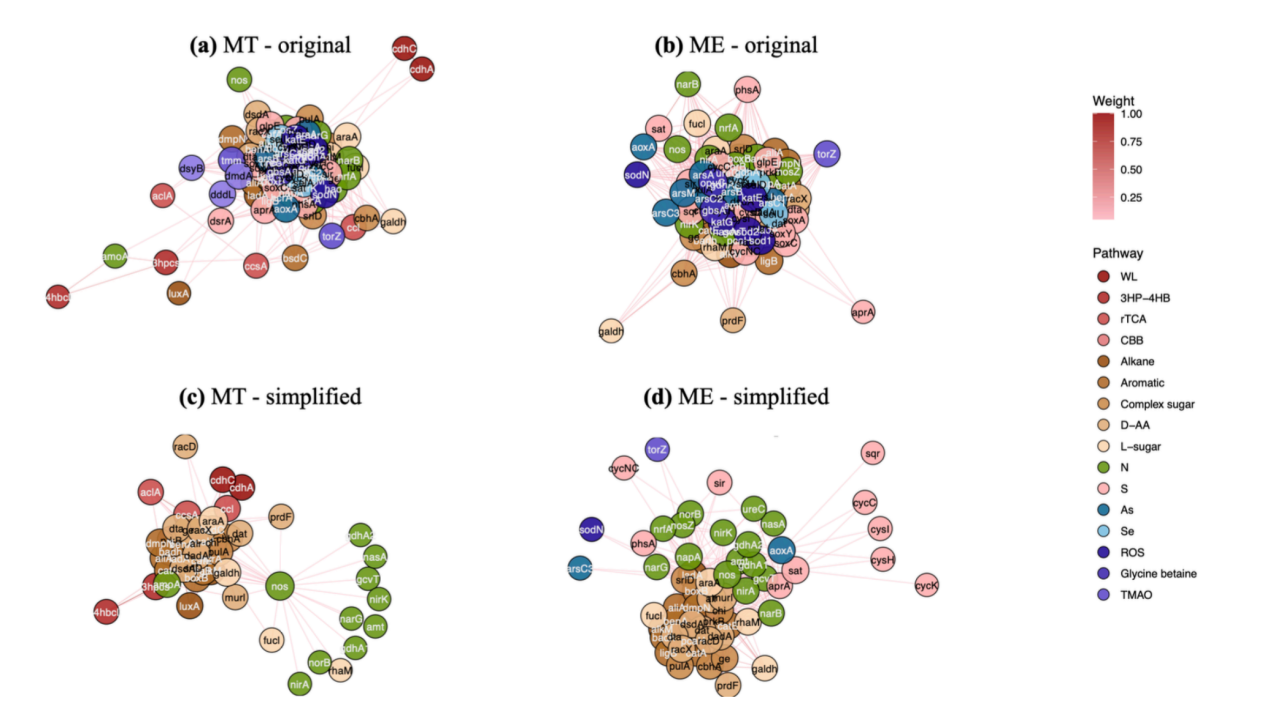
Wir haben zuvor Gen-Koexistenznetzwerke verwendet, um Studien zur Vereinfachung sozialer Netzwerke durchzuführen. Wie in der Abbildung unten gezeigt, sind Abbildung a und b unterschiedliche Netzwerke desselben Gens aus der Tiefsee und dem Hochgebirge. Ihre ursprünglichen Formen sind komplex und unorganisiert. Durch die Konstruktion eines Graph-Neuralnetzwerks, das dem menschlichen sozialen Netzwerk ähnelt, vereinfachen wir die beiden Netzwerke, identifizieren die Gene, die eine absolute dominante Stellung einnehmen, und unterscheiden, welche Gene engere Verbindungen aufweisen und welche Gene relativ schwache Verbindungen haben. Das vereinfachte Netzwerk kann Biologen dabei helfen, ihr Fachwissen zur Analyse von Netzwerken und biologischen Gemeinschaften zu nutzen. Eine vorläufige Version der Studie trägt den Titel „Eine einheitliche Sicht auf die neuronale Nachrichtenübermittlung mit Meinungsdynamik für soziale Netzwerke.“
Adresse des Vorabdrucks:
https://arxiv.org/abs/2310.01272
Das zweite Beispiel ist die Interpretierbarkeitsforschung auf Basis eines Graph-Neural-Networks. Ein intuitives Beispiel ist, dass Graph-Neuronale Netzwerke dabei helfen können, wichtige lokale Strukturen innerhalb von Molekülen zu identifizieren. Dieses Ergebnis kann einerseits dazu genutzt werden, die Rationalität des Modells zu testen. Wenn das Modell beispielsweise bei der Vorhersage der Proteinfunktion Schlüsselatome oder Aminosäuren bis zu einem gewissen Grad in der Nähe des aktiven Zentrums lokalisieren kann, bedeutet dies, dass das Modell über eine gewisse Rationalität verfügt. Wenn die Aufmerksamkeit des Modells hingegen zufällig und diskret auf mehrere Aminosäuren auf der Proteinoberfläche verteilt ist, kann es zu Problemen mit dem Modell kommen. Andererseits könnte ein sinnvolles und leistungsfähiges Erklärungsmodell, das die Rolle jedes Knotens bei der funktionellen Vorhersage analysiert, im Idealfall in Zukunft sogar dabei helfen, Taschenregionen völlig neuer Proteine zu identifizieren.
Obwohl große Modelle in vielen Anwendungsszenarien umfangreiche erfolgreiche Erfahrungen geliefert haben, sind sie nicht die einzige Lösung für alle Probleme. Als Bereich, in dem natürlicherweise verschiedene strukturierte Daten existieren, können Graph-Neural-Networks mögliche Lösungen für viele Probleme in der Biologie bieten. Unabhängig davon, ob es sich um Moleküle, Komplexe, Gene, mikrobielle Netzwerke oder größere und komplexere Systeme handelt, können Graph-Neural-Networks eine einfache Lösung bieten, indem sie induktive Verzerrungen einbauen und das menschliche Vorwissen auch bei kleinen Datenmengen maximieren.
Über Zhou Bingxin

Zhou Bingxin ist derzeit Assistenzforscher am National Center for Applied Mathematics (Shanghai Jiao Tong University). Sie erhielt ihren Doktortitel 2022 von der University of Sydney, Australien, und war Gastwissenschaftlerin an der University of Cambridge, Großbritannien. Sein Forschungsschwerpunkt liegt auf der Nutzung von Deep Learning (insbesondere geometrischem Deep Learning) zur Lösung biologischer Herausforderungen, wie etwa Enzym-Engineering, metabolische Gennetzwerke und die Analyse der Evolution von Proteinstrukturgruppen. Die entwickelten Deep-Learning-Algorithmen werden zur Verarbeitung statischer, dynamischer, heterogener und verrauschter Graphen verwendet und einige von ihnen wurden in führenden internationalen Fachzeitschriften und Konferenzen wie IEEE TPAMI, JMLR, ICML und NeurIPS veröffentlicht. Das allgemeine Deep-Learning-Framework für Protein-Engineering und Sequenzdesign kann die Aktivität komplexer Proteine effektiv gestalten und deutlich verbessern. Einige der Ergebnisse wurden in Zeitschriften wie eLife und Chem veröffentlicht. Sci. und ACS JCIM.
Persönliche Homepage:
https://ins.sjtu.edu.cn/peoples/ZhouBingxin
Google Scholar:








