Command Palette
Search for a command to run...
Ausgewählt Für ICML! Das Team Der Renmin-Universität Nutzte Das Äquivariante Graph-Neuronale Netzwerk Zur Vorhersage Von Zielprotein-Bindungsstellen, Wobei Die Größte Leistungsverbesserung Bei 20% Zu Verzeichnen war.
In lebenden Systemen sind bei fast allen biologischen und pharmakologischen Prozessen Wechselwirkungen zwischen Rezeptoren (Zielproteinen) und Liganden (kleinen Molekülen) beteiligt. Diese Wechselwirkungen treten in bestimmten Regionen der Zielproteinstruktur auf.Die Vorhersage der Bindungsstellen von Zielproteinen, auch als „Bindungsstellen“ bekannt, spielt eine grundlegende Rolle bei verschiedenen nachgelagerten Aufgaben, beispielsweise bei der Arzneimittelforschung.
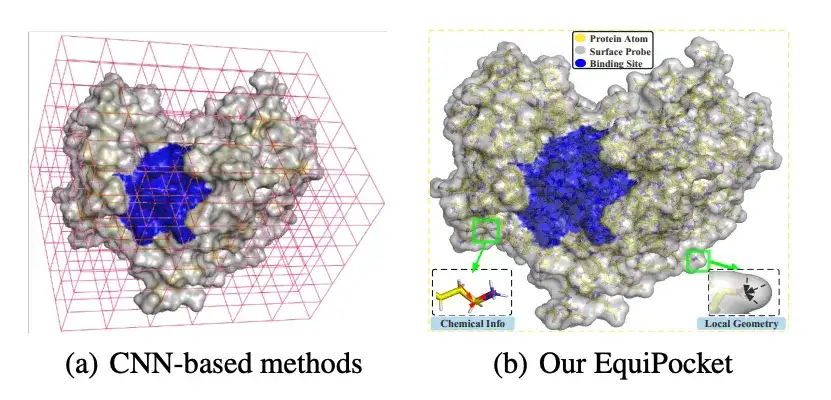
In den letzten Jahren wurden Convolutional Neural Networks (CNNs) – inspiriert durch den Durchbruch des Deep Learning – erfolgreich zur Vorhersage von Ligandenbindungsstellen eingesetzt. CNN-basierte Methoden behandeln Proteine als 3D-Bilder, indem sie die Atome der Proteine räumlich in den nächstgelegenen Voxeln gruppieren und dann die Vorhersage der Bindungsstelle als Objekterkennungsproblem oder semantische Segmentierungsaufgabe auf einem 3D-Gitter modellieren. Diese Methoden haben gewisse Vorteile, es gibt aber immer noch Herausforderungen, wie zum BeispielEs weist Mängel bei der Darstellung unregelmäßiger Proteinstrukturen auf; es ist rotationsempfindlich; es beschreibt die geometrischen Merkmale von Proteinoberflächen nicht ausreichend; und es ist unempfindlich gegenüber Änderungen der Proteingröße.
Zu diesem Zweck veröffentlichte ein Forschungsteam der Gaoling School of Artificial Intelligence der Renmin University of China kürzlich auf der ICML 2024, der wichtigsten akademischen Konferenz im Bereich der KI, ein Forschungspapier mit dem Titel „EquiPocket: ein E(3)-äquivariantes geometrisches Graph-Neuralnetzwerk zur Vorhersage von Ligandenbindungsstellen“. In dieser Studie wird erstmals ein E(3)-isovariantes Graph-Neural-Netzwerk (GNN) zur Vorhersage von Ligandenbindungsstellen eingesetzt.Vorgeschlagen wurde ein Framework namens EquiPocket,Die Herausforderungen, denen CNN-basierte Methoden gegenüberstehen, werden angesprochen.
Forschungshighlights:
* Die erste Anwendung von E(3) isovariantem GNN zur Vorhersage von Ligandenbindungsstellen
* Im Vergleich zu herkömmlichen CNN-basierten Methoden erfordert EquiPocket keine Voxelisierung, kann unregelmäßige Proteinstrukturen modellieren und ist unempfindlich gegenüber euklidischen Transformationen. Dadurch werden Herausforderungen wie „Defekte bei der Darstellung unregelmäßiger Proteinstrukturen“ und „Rotationsempfindlichkeit“ gelöst.
* Umfangreiche Experimente mit repräsentativen Benchmark-Methoden zeigen die Überlegenheit von EquiPocket gegenüber den aktuellen State-of-the-Art-Methoden, was für verschiedene nachgelagerte Aufgaben wie die Arzneimittelforschung hilfreich sein kann.
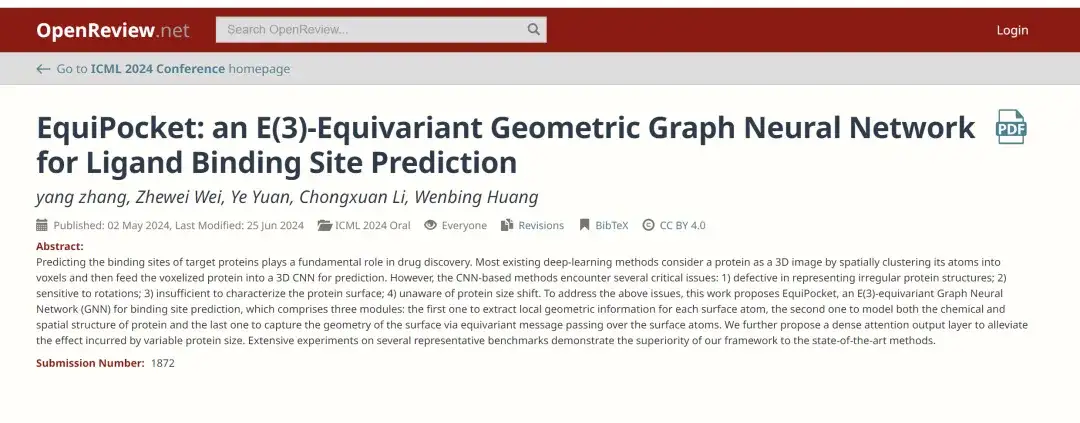
Papieradresse:
https://openreview.net/forum?id=1vGN3CSxVs
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Umfassende Überprüfung mehrerer professioneller Datensätze
Die Forscher wählten mehrere spezialisierte Datensätze aus und werteten sie mithilfe einer Teilmenge von mlig aus jedem Datensatz aus, die relevante Liganden für die Vorhersage von Bindungsstellen enthielt.
In,scPDB ist ein bekannter Datensatz zur Vorhersage von Bindungsstellen.Enthält von VolSite generierte Protein-, Liganden- und 3D-Hohlraumstrukturen. In dieser Studie wurde die Version 2017 zum Training und zur Kreuzvalidierung verwendet, die 17.594 Strukturen, 16.034 Einträge, 4.782 Proteine und 6.326 Liganden enthält.
PDBbind ist ein häufig verwendeter Datensatz zum Studium von Protein-Liganden-Komplexen.Enthält die 3D-Struktur des Proteins, des Liganden, der Bindungsstelle und genaue, im Labor ermittelte Ergebnisse der Bindungsaffinität. In dieser Studie wurde zur Auswertung die Version von 2020 verwendet, die aus zwei Teilen besteht: einem gemeinsamen Satz (14.127 Komplexe) und einem verfeinerten Satz (5.316 Komplexe). Der allgemeine Satz enthält alle Protein-Liganden-Komplexe und der verfeinerte Satz wählt Verbindungen mit besserer Qualität aus dem allgemeinen Satz für experimentelle Tests aus.
COACH 420 und HOLO4K sind zwei Testdatensätze, die zur Vorhersage von Bindungsstellen verwendet werden.Erstmals vorgestellt von (Krivák & Hoksza, 2018).
Modellarchitektur: Das Gesamtframework von EquiPocket besteht aus drei Modulen
Das Gesamtgerüst von EquiPocket besteht aus 3 Modulen:Wie in der folgenden Abbildung dargestellt:
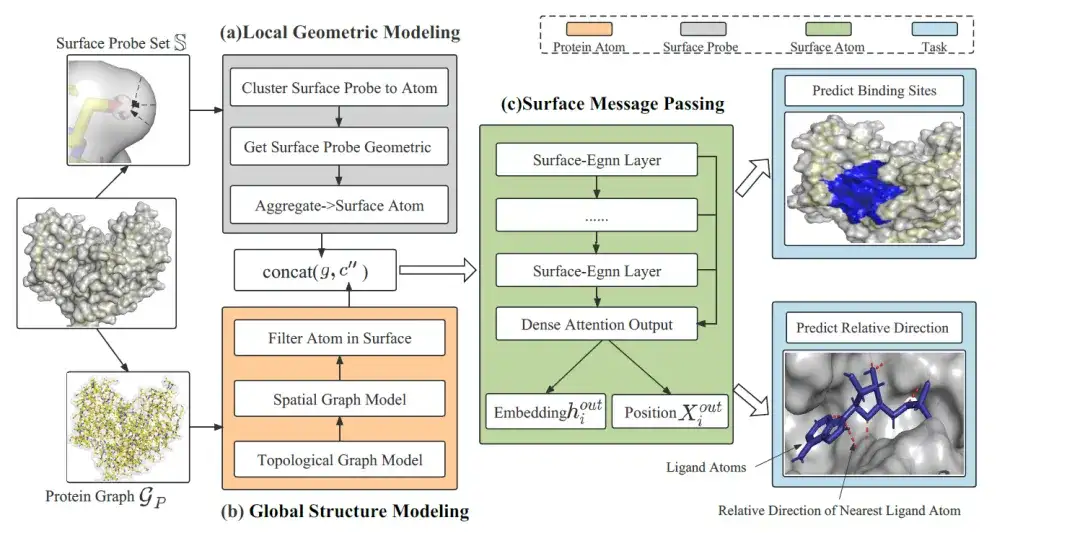
Das erste Modul ist das Modul „Local Geometric Modeling“, das zum Extrahieren der lokalen geometrischen Informationen jedes Oberflächenatoms verwendet wird. Das zweite Modul ist das Modul „Global Structure Modeling“, das zur Beschreibung der chemischen und räumlichen Struktur von Proteinen verwendet wird. Das letzte Modul ist das Surface Message Passing-Modul, das die Oberflächengeometrie erfasst, indem es äquivariante Informationen zu Oberflächenatomen überträgt.
Modul zur lokalen Geometriemodellierung
Die lokale Geometrie jedes Proteinatoms bestimmt, ob die benachbarte Region geeignet ist, Teil der Bindungsstelle zu sein.
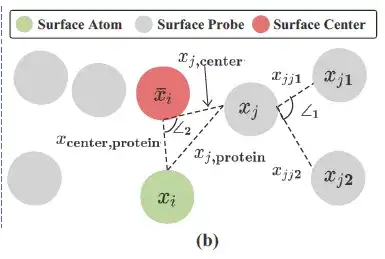
Wie in der Abbildung oben gezeigt, verwendeten die Forscher Oberflächensonden (grau in der Abbildung oben) um jedes Proteinoberflächenatom (Oberflächenatom, grün in der Abbildung oben), um die lokalen geometrischen Informationen zu beschreiben. Insbesondere werden für jedes Oberflächenatom i ∈ VS die Oberflächensonden um es herum von einer Teilmenge von S zurückgegeben, und zwar:
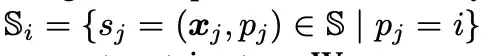
Die Forscher konstruierten geometrische Informationen auf der Grundlage von Si und zeichneten den Mittelpunkt/Durchschnittswert aller 3D-Koordinaten in Si (Oberflächenmittelpunkt, rot in der Abbildung oben) als xi¯ auf.
Globales Strukturmodellierungsmodul
Obwohl Bindungsstellen hauptsächlich aus Oberflächenatomen bestehen, beeinflusst die Gesamtstruktur des Proteins häufig die Ligandeninteraktionen sowie die Bildung von Bindungsstellen und muss daher modelliert werden.
Die Forscher erreichten dieses Ziel durch zwei miteinander verbundene Prozesse: chemische Graphenmodellierung und räumliche Graphenmodellierung. Das resultierende globale Strukturmodellierungsmodul ist für die Verarbeitung von Informationen über das gesamte Protein verantwortlich, einschließlich Atomtypen, chemischen Bindungen, relativen räumlichen Positionen usw.
Modul zur Übertragung von Oberflächeninformationen
Unter Berücksichtigung der lokalen geometrischen Merkmale der Oberflächenatome und der globalen Kodierungsmerkmale führt dieses Modul eine äquivariante Informationsübertragung auf der Oberflächenkarte durch, um alle Merkmale der Proteinoberflächenatome zu aktualisieren.
Forschungsergebnisse: EquiPocket verbessert die Leistung um 10-20% im Vergleich zu Basismodellen
Im Experiment verglichen die Forscher EquiPocket mit den folgenden Basismodellen:
* Geometriebasierte Methoden: Fpocket
* Maschinelles Lernverfahren: P2rank
* CNN-basierte Methoden: DeepSite, Kalasanty, DeepSurf, RecurPocket
* Topologiebasierte Modelle: GAT, GCN und GCN2
* Räumliche graphenbasierte Modelle: SchNet, EGNN
Zu den zur Bewertung des Modells verwendeten Metriken gehören DCC (der Abstand zwischen dem vorhergesagten Mittelpunkt der Bindungsstelle und dem wahren Mittelpunkt der Bindungsstelle), DCA (der kürzeste Abstand zwischen dem vorhergesagten Mittelpunkt der Bindungsstelle und einem beliebigen Ligandenraster) und die Ausfallrate (die Abtastrate ohne vorhergesagten Mittelpunkt der Bindungsstelle). Die folgende Tabelle zeigt die Ergebnisse der Bindungsstellenvorhersage auf COACH 420, HOLO4K und PDBbind.
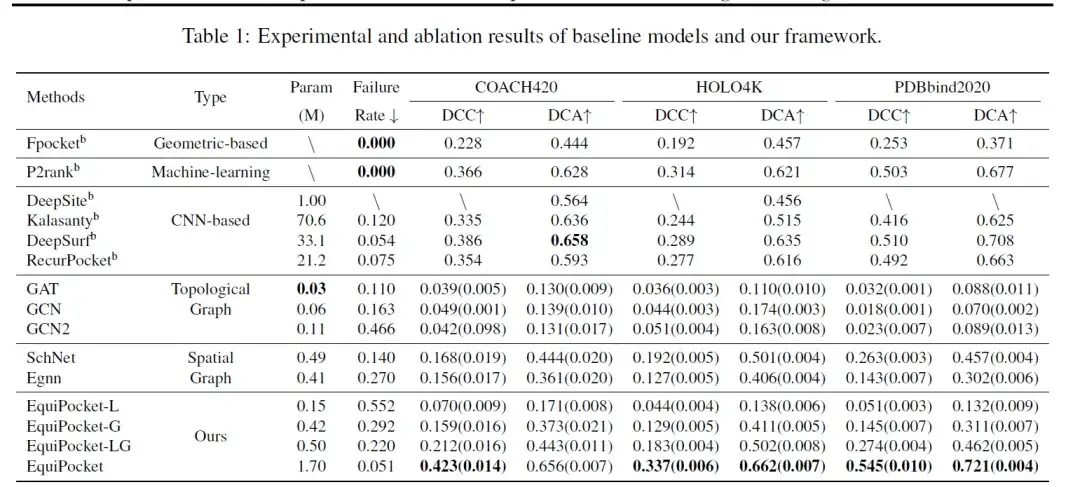
Wie aus den Daten in der Tabelle hervorgeht,Die geometriebasierte Methode Fpocket weist eine schlechte Leistung auf.Da diese Methode nur die geometrischen Merkmale des Proteins verwendet, beträgt die Fehlerrate 0; Die Machine-Learning-Methode P2rank kombiniert Random Forests mit den geometrischen Informationen der Proteinoberfläche und verbessert dadurch die Performance deutlich.
CNN-basierte Methoden (DeepSite, Kalasanty, DeepSurf, RecurPocket) weisen eine wesentlich bessere Leistung auf als geometriebasierte Methoden.Unter ihnen stellen DCC und DCA eine größere Verbesserung als 50% dar, erfordern jedoch eine große Anzahl von Parametern und Rechenressourcen. Unter diesen sind die früher vorgeschlagenen Methoden DeepSite und Kalasanty durch die Variation der Proteingröße und die unzureichende Fähigkeit, große Proteine zu verarbeiten, eingeschränkt, was zu Vorhersagefehlern führen kann.
Für Graphenmodelle:Die Leistung topologischer Graphenmodelle (GCN, GAT, GCN2) ist schlecht.Dies liegt hauptsächlich daran, dass sie nur Informationen zu Atom- und chemischen Bindungen berücksichtigen und die räumliche Struktur von Proteinen ignorieren. Die Leistung räumlicher Graphenmodelle (SchNet, EGNN) ist normalerweise besser als die topologischer Graphenmodelle. EGNN verwendet die Eigenschaften von Atomen sowie ihre relativen/absoluten räumlichen Positionen, was besser funktioniert; SchNet aktualisiert Einbettungen nur auf Grundlage der relativen Abstände von Atomen, die Leistung räumlicher Graphenmodelle ist jedoch schlechter als die auf CNN und Geometrie basierender Methoden, da erstere keine ausreichenden geometrischen Merkmale erhalten und das Problem der Größenänderungen von Proteinen nicht lösen können.
Die obigen Ergebnisse zeigen, dassDie geometrischen Informationen und die mehrstufigen Strukturinformationen der Proteinoberfläche sind für die Vorhersage der Bindungsstelle von entscheidender Bedeutung.
Darüber hinaus spiegelt dies auch die Einschränkungen des aktuellen GNN-Modells wider, d. h., es ist schwierig, ausreichend geometrische Informationen von der Proteinoberfläche zu sammeln, oder die erforderlichen Rechenressourcen sind zu groß, was die Anwendung auf makromolekulare Systeme wie Proteine erschwert. Daher kann das EquiPocket-Framework nicht nur chemische und räumliche Informationen auf atomarer Ebene aktualisieren, sondern auch effizient geometrische Informationen sammeln, ohne dass übermäßige Rechenressourcen erforderlich sind.Seine Leistung verbessert die vorherigen Ergebnisse um 10-20%.
Von kleinen Molekülliganden bis hin zu biologischen Makromolekülen: KI interpretiert Proteinstrukturen detailliert
In jeder pflanzlichen, tierischen und menschlichen Zelle gibt es Milliarden molekularer Maschinen, die aus Molekülen wie Proteinen, Nukleinsäuren und Zuckern bestehen, und kein einzelner Teil davon kann für sich allein funktionieren. Nur wenn wir verstehen, wie sie in Millionen von Kombinationen interagieren, können wir ein tieferes Verständnis des Lebens gewinnen.
Im Mai dieses Jahres veröffentlichte Google DeepMind das Modell AlphaFold3, das gemeinsame Strukturvorhersagen für Komplexe wie Proteine, Nukleinsäuren, kleine Moleküle, Ionen und modifizierte Rückstände durchführen kann. Die Interaktion zwischen Proteinen und kleinen Molekülliganden ist der Kern des Wirkmechanismus des Medikaments. Durch seinen fortschrittlichen Deep-Learning-Algorithmus kann AlphaFold3 die dreidimensionale Struktur der Protein-Liganden-Bindung präzise vorhersagen, und zwar mit einer Genauigkeit, die die vorhandener Docking-Tools bei weitem übertrifft.
Im Hinblick auf die Entwicklung neuer MedikamenteDurch die von AlphaFold3 vorhergesagten Protein-Liganden-Strukturen können Forscher neue Arzneimittelkandidaten effektiver prüfen und entwickeln und den Prozess der Arzneimittelentdeckung beschleunigen. Im Hinblick auf die Optimierung bestehender Medikamente kann dieses Tool auch zur Optimierung bestehender Medikamente verwendet werden, indem ihre Bindungsart mit dem Zielprotein verbessert wird, um die Wirksamkeit zu erhöhen oder Nebenwirkungen zu verringern.
Zusätzlich zu kleinen Molekülliganden,Um ihre biologischen Funktionen erfüllen zu können, müssen Proteine außerdem mit biologischen Makromolekülen wie DNA und Zuckern interagieren.Mittlerweile sind durch experimentelle Methoden tausende Proteinstrukturkomplexe in der Proteindatenbank hinterlegt. Herkömmliche experimentelle Methoden sind jedoch zeitaufwändig und teuer, während auf maschinellem Lernen basierende Vorhersagemethoden die Herausforderungen problemlos lösen können.
Im Februar dieses Jahres veröffentlichte ein Forschungsteam der Nanjing Agricultural University eine Online-Forschungsarbeit mit dem Titel „ULDNA: Integration unüberwachter Multi-Source-Sprachmodelle mit LSTM-Attention Network für die hochgenaue Vorhersage von Protein-DNA-Bindungsstellen“ in Briefings in Bioinformatics, einer wichtigen Zeitschrift auf dem Gebiet der Biologie.Zur Vorhersage von Protein-DNA-Bindungsstellen wurde eine neue Deep-Learning-Vorhersagemethode (ULDNA) entwickelt.
Papieradresse:
https://academic.oup.com/bib/article/25/2/bbae040/7606634

Die Kernidee von ULDNA besteht darin, das Proteinsprachenmodell zu verwenden, um die Merkmalsdarstellung für die Sequenz zu entwerfen, und dann das Langzeit-Kurzzeitgedächtnisnetzwerk (LSTM-Attention Network) mit dem Aufmerksamkeitsmechanismus zu kombinieren, um das Vorhersagemodell für DNA-Bindungsstellen zu trainieren. Die Forscher wählten sieben Benchmark-Datensätze aus, darunter PDNA-128, PDNA-316 und PDNA-335 (mit einer Anzahl von Proteinsequenzen zwischen 40 und 600), und führten einen umfassenden Test an ULDNA durch. Die experimentellen Ergebnisse zeigen, dassULDNA liefert bei allen Datensätzen gute Ergebnisse und seine Vorhersageleistung ist deutlich besser als die der anderen neun gängigen Methoden.
Zusätzlich zur DNA sind Zucker auf den Zelloberflächen aller Organismen allgegenwärtig, wo sie mit zahlreichen Proteinfamilien wie Lektinen, Antikörpern, Enzymen und Transportern interagieren, um wichtige biologische Prozesse wie Immunreaktionen, Zelldifferenzierung und neuronale Entwicklung zu regulieren.Das Verständnis der Wechselwirkungsmechanismen zwischen Kohlenhydraten und Proteinen ist die Grundlage für die Entwicklung von Kohlenhydratmedikamenten.Allerdings stellen die Vielfalt und Komplexität der Kohlenhydratstrukturen, insbesondere die Variabilität ihrer Proteinbindungsstellen, eine Herausforderung für die Erfassung experimenteller Daten und die Arzneimittelentwicklung dar.
Erst kürzlich hat ein Team der Chinesischen Akademie der Wissenschaften ein Deep-Learning-Modell namens DeepGlycanSite entwickelt, das Zuckerbindungsstellen auf einer gegebenen Proteinstruktur genau vorhersagen kann. DeepGlycanSite integriert die geometrischen und evolutionären Merkmale von Proteinen in ein tiefes äquivariantes Graph-Neuralnetzwerk, das auf der Transformer-Architektur basiert.Seine Leistung übertrifft frühere fortschrittliche Methoden erheblich und ermöglicht eine effektive Vorhersage der Bindungsstellen verschiedener Zuckermoleküle.
In Kombination mit Mutagenesestudien enthüllte DeepGlycanSite eine Guanosin-5'-diphosphat-Zuckererkennungsstelle für einen wichtigen G-Protein-gekoppelten Rezeptor. Diese Erkenntnisse legen nahe, dass DeepGlycanSite für die Vorhersage von Zuckerbindungsstellen wertvoll ist und Einblicke in die molekularen Mechanismen hinter der Kohlenhydratregulierung therapeutisch wichtiger Proteine liefern kann.

Die Studie mit dem Titel „Hochpräzise Vorhersage von Kohlenhydrat-Bindungsstellen mit DeepGlycanSite“ wurde am 17. Juni 2024 in Nature Communications veröffentlicht.
Papieradresse:
https://www.nature.com/articles/s41467-024-49516-2
Zusammenfassend lässt sich sagen, dass Proteine wichtige Moleküle in lebenden Organismen sind und eine Schlüsselrolle bei der Struktur und Funktion von Zellen spielen. Die Untersuchung der Proteinstruktur ist für das Verständnis von Lebensprozessen, die Aufdeckung von Krankheitsmechanismen und die Entwicklung von Medikamenten von großer Bedeutung. Heute öffnet maschinelles Lernen den Wissenschaftlern neue Türen zum Verständnis der Geheimnisse des Lebens.
Quellen:
1.https://openreview.net/forum?id=1vGN3CSxVs
2.https://mp.weixin.qq.com/s/aGzcr0ncQA-jBy-vTGC35Q
3.https://www.jiqizhixin.com/articles/2024-05-09
4.https://news.njau.edu.cn/2024/0








