Command Palette
Search for a command to run...
Neuronale Netze Ersetzen Die Dichtefunktionaltheorie! Tsinghua-Forschungsgruppe Veröffentlicht Universelles Materialmodell DeepH Und Erreicht Damit Ultragenaue Vorhersage

Beim Materialdesign ist das Verständnis der elektronischen Struktur und Eigenschaften der Schlüssel zur Vorhersage der Materialleistung, zur Entdeckung neuer Materialien und zur Optimierung der Materialleistung. Vergangenheit,Die Dichtefunktionaltheorie (DFT) wird in der Industrie häufig zur Untersuchung der elektronischen Struktur und Eigenschaften von Materialien eingesetzt. Sein Wesen besteht darin, die Elektronendichte als Träger aller Informationen im Grundzustand von Molekülen (Atomen) zu verwenden.Anstelle der Wellenfunktion eines einzelnen Elektrons wird das Mehrelektronensystem zur Lösung in ein Einzelelektronenproblem umgewandelt, was nicht nur den Berechnungsprozess vereinfacht, sondern auch die Berechnungsgenauigkeit gewährleistet und die Aperturverteilung genauer wiedergeben kann.
Allerdings ist die DFT rechenintensiv und kann normalerweise nur zur Untersuchung kleiner Materialsysteme verwendet werden. Inspiriert von der Materials Genome Initiative haben Wissenschaftler begonnen, mithilfe von DFT eine riesige Materialdatenbank aufzubauen. Obwohl bisher nur begrenzte Datensätze gesammelt wurden, ist dies bereits ein bemerkenswerter Anfang. Mit diesem Ausgangspunkt begannen die Forscher angesichts neuer Veränderungen durch die KI-Technologie darüber nachzudenken: „Kann die Kombination von Deep Learning mit DFT und die Möglichkeit, neuronalen Netzwerken das Wesentliche von DFT tiefgreifend zu erlernen, einen revolutionären Durchbruch bringen?“
Dies ist der Kern der Deep Learning Density Functional Theory Hamiltonian (DeepH)-Methode.Durch die Einkapselung der Komplexität der DFT in ein neuronales Netzwerk ist DeepH nicht nur in der Lage, Berechnungen mit beispielloser Geschwindigkeit und Effizienz durchzuführen, sondern seine Intelligenz verbessert sich auch kontinuierlich, wenn weitere Trainingsdaten hinzugefügt werden.Kürzlich hat die Forschungsgruppe von Xu Yong und Duan Wenhui vom Institut für Physik der Tsinghua-Universität ihre ursprüngliche DeepH-Methode erfolgreich zur Entwicklung des universellen DeepH-Materialmodells eingesetzt und eine praktikable Lösung für den Aufbau eines „großen Materialmodells“ demonstriert. Dieser Durchbruch bietet neue Möglichkeiten für die Entdeckung innovativer Materialien.
Die zugehörige Forschung mit dem Titel „Universal materials model of deep-learning density functional theory Hamiltonian“ wurde im Science Bulletin veröffentlicht.

Papieradresse:
https://doi.org/10.1016/j.scib.2024.06.011
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Erstellen Sie mit AiiDA eine große Materialdatenbank, um magnetische Materialinterferenzen zu eliminieren
Um die Universalität des allgemeinen DeepH-Materialmodells zu demonstrieren, wurde im Rahmen dieser Studie mithilfe der Automatic Interactive Infrastructure and Database (AiiDA) eine große Materialdatenbank mit 104 festen Materialien erstellt.
Um die vielfältige Elementzusammensetzung aufzuzeigen, wurden in der Studie auch die ersten vier Reihen des Periodensystems ausgewählt. Dabei wurden Übergangselemente von Sc bis Ni ausgeschlossen, um Störungen durch magnetische Materialien zu vermeiden, und Edelgaselemente wurden ebenfalls ausgeschlossen. Die in Frage kommenden Materialstrukturen stammen aus der Materials Project-Datenbank. Zusätzlich zur Filterung nach Elementtyp werden in der Spalte „Materialien“ die Kandidatenmaterialien weiter verfeinert, um nur die als „nicht magnetisch“ gekennzeichneten einzuschließen. Der Einfachheit halber wurden Strukturen mit mehr als 150 Atomen in der Elementarzelle ausgeschlossen.
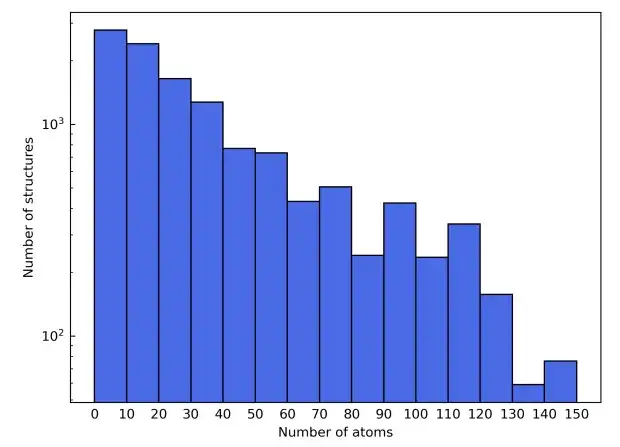
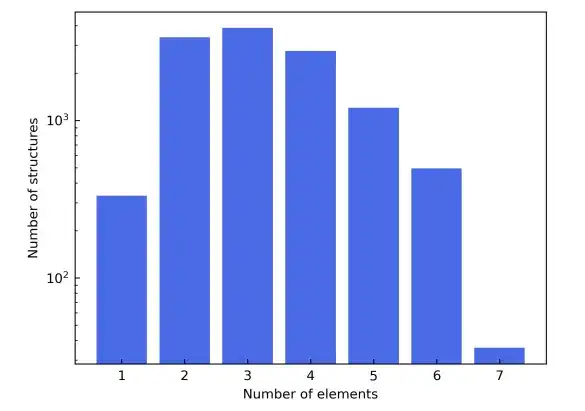
Als Ergebnis dieser Filterkriterien bestand der endgültige Materialdatensatz aus insgesamt 12.062 Strukturen. Während des Trainingsprozesses wird der Datensatz im Verhältnis 6:2:2 in Trainingssatz, Validierungssatz und Testsatz aufgeteilt. Nächste,Die Studie verwendete das AiiDA-Framework (Automated Interactive Infrastructure and Database), um einen Hochdurchsatz-Workflow für Dichtefunktionaltheorie-Berechnungen zu entwickeln und nutzte es zum Aufbau einer Materialdatenbank.
Mit dem DFT-Hamiltonoperator als Ziel wird DeepH mit der DeepH-2-Methode trainiert.
Untersuchungen legen nahe, dassDer DFT-Hamiltonoperator ist ein ideales Ziel für maschinelles Lernen.
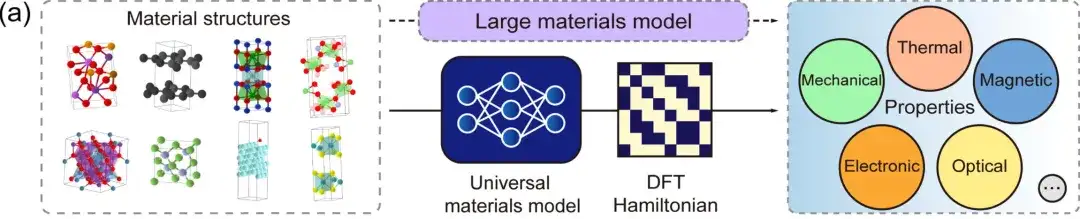
Erste,Der DFT-Hamiltonoperator ist eine grundlegende Größe, die direkt aus physikalischen Größen wie Gesamtenergie, Ladungsdichte, Bandstruktur und physikalischen Reaktionen abgeleitet werden kann.Das allgemeine DeepH-Materialmodell kann jede Materialstruktur als Eingabe akzeptieren und den entsprechenden DFT-Hamiltonoperator generieren, sodass verschiedene Materialeigenschaften direkt abgeleitet werden können, wie in der obigen Abbildung gezeigt.
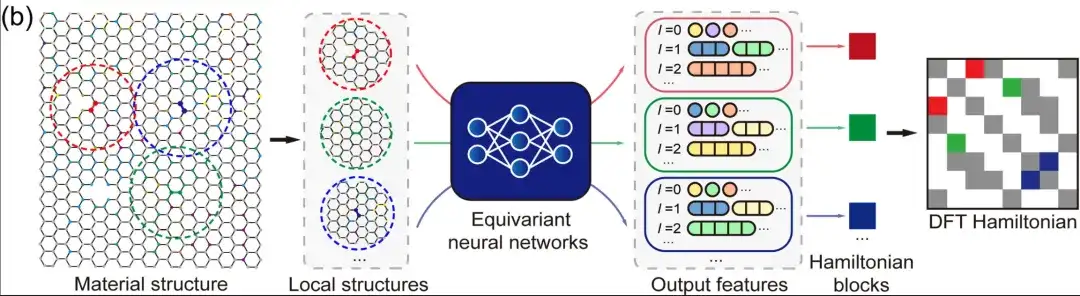
Zweitens,Im Fall eines lokalisierten atomaren Basissatzes kann der DFT-Hamiltonoperator als dünn besetzte Matrix dargestellt werden, deren Elemente durch die lokale chemische Umgebung bestimmt werden.In äquivarianten neuronalen Netzwerken verwendet DeepH Ausgabemerkmale, die mit unterschiedlichen Winkelquantenzahlen l gekennzeichnet sind, um den DFT-Hamiltonoperator darzustellen, wie in der obigen Abbildung gezeigt. Daher kann man die Hamilton-Matrixelemente zwischen Atompaaren basierend auf den benachbarten Strukturinformationen modellieren, ohne die DFT-Hamilton-Matrix der gesamten Materialstruktur modellieren zu müssen. Dies vereinfacht nicht nur die Deep-Learning-Aufgabe erheblich, sondern erhöht auch die Menge der Trainingsdaten erheblich. Was die Inferenz betrifft, lässt sich das trainierte Modell gut auf neue, bisher unbekannte Materialstrukturen verallgemeinern, sobald das Deep-Learning-Netzwerk genügend Trainingsdaten gelernt hat.
Die Kernidee von DeepH besteht darin, neuronale Netzwerke zur Darstellung von HDFT zu verwenden.Durch Variation der Eingangsmaterialstruktur werden zunächst durch den DFT-Code generierte HDFT-Trainingsdaten erstellt, die dann zum Trainieren des neuronalen Netzes verwendet werden. Diese trainierten Netzwerkmodelle werden dann verwendet, um auf neue Materialstrukturen zu schließen.
Dabei sind zwei wichtige Vorkenntnisse wichtig: das Lokalitätsprinzip,Diese Studie stellt den DFT-Hamiltonoperator in einer lokalisierten Atomprobe dar und zerlegt den Hamiltonoperator in Blöcke, die entweder interatomare oder intraatomare Kopplungen beschreiben. Daher kann eine einzelne Trainingsmaterialstruktur einem Hamilton-Block für eine große Datenmenge entsprechen. Darüber hinaus kann jeder Hamiltonblock anhand von Informationen über die lokale Struktur und nicht anhand der gesamten Struktur bestimmt werden. Diese Vereinfachung gewährleistet die hohe Genauigkeit und Übertragbarkeit des DeepH-Modells.
Das zweite ist das Prinzip der Symmetrie.Die Gesetze der Physik bleiben gleich, wenn man sie aus verschiedenen Koordinatensystemen betrachtet. Daher sind die entsprechenden physikalischen Größen und Gleichungen bei Koordinatentransformation äquivalent. Durch die Beibehaltung der Äquivalenz wird nicht nur die Dateneffizienz verbessert, sondern auch die Generalisierungsfähigkeit gesteigert, was die Leistung von DeepH erheblich steigern kann. Die DeepH-Architektur der ersten Generation vereinfacht das Äquivalenzproblem durch ein lokales Koordinatensystem und stellt äquivalente Merkmale durch Transformation lokaler Koordinaten wieder her. Die DeepH-Architektur der zweiten Generation basiert auf einem gleichwertigen neuronalen Netzwerk und trägt den Namen DeepH-E3. In diesem Rahmen sind die Merkmalsvektoren aller Eingabe-, verborgenen und Ausgabeebenen äquivalente Vektoren. Vor Kurzem hat einer der Autoren dieser Arbeit eine Architektur der neuen Generation für Deep Learning vorgeschlagen: DeepH-2. In Bezug auf Effizienz und Genauigkeit schneidet DeepH-2 am besten ab.
Zusammenfassend wurde das Deep-Learning-Modell DeepH dieser Studie mit der DeepH-2-Methode trainiert und enthält insgesamt 17,28 Millionen Parameter. Es bildete ein neuronales Netzwerk, das für die Nachrichtenübermittlung basierend auf 3 äquivalenten Transformationsblöcken verwendet werden kann, und jeder Knoten und jede Kante trug 80 äquivalente Merkmale.Die Materialstruktur ist mit der Ordnungszahl und dem interatomaren Abstand eingebettet und es wird eine Gaußsche Glättungsstrategie angewendet, wobei der mittlere Bereich der Basisfunktionen von 0,0 bis 9,0 Å reicht. Die Ausgabefunktionen des neuronalen Netzwerks werden durch die lineare Schicht geleitet und dann wird der DFT-Hamiltonoperator durch die Wigner-Eckart-Schicht erstellt.
Die Studie wurde 343 Epochen und 207 Stunden lang auf NVIDIA A100-GPUs trainiert. Während des gesamten Trainingsprozesses ist die Batchgröße auf 1 festgelegt, was bedeutet, dass jeder Batch eine Materialstruktur enthält. Schließlich beträgt die anfängliche Lernrate 4 × 10-4, die Abklingrate 0,5, die Abklinggeduld 20, die minimal ausgewählte Lernrate 1 × 10-5 und das Training wird gestoppt, wenn die Lernrate diesen Wert erreicht.
DeepH verfügt über eine hervorragende Inferenzleistung und kann genaue Bandstrukturvorhersagen liefern
Die mittleren absoluten Fehler (MAE) der vom Modell für die Trainings-, Validierungs- und Testsätze vorhergesagten Hamilton-Matrixelemente der Dichtefunktionaltheorie betrugen 1,45, 2,35 bzw. 2,20 meV.Dies zeigt die Fähigkeit des Modells, über unsichtbare Strukturen nachzudenken.
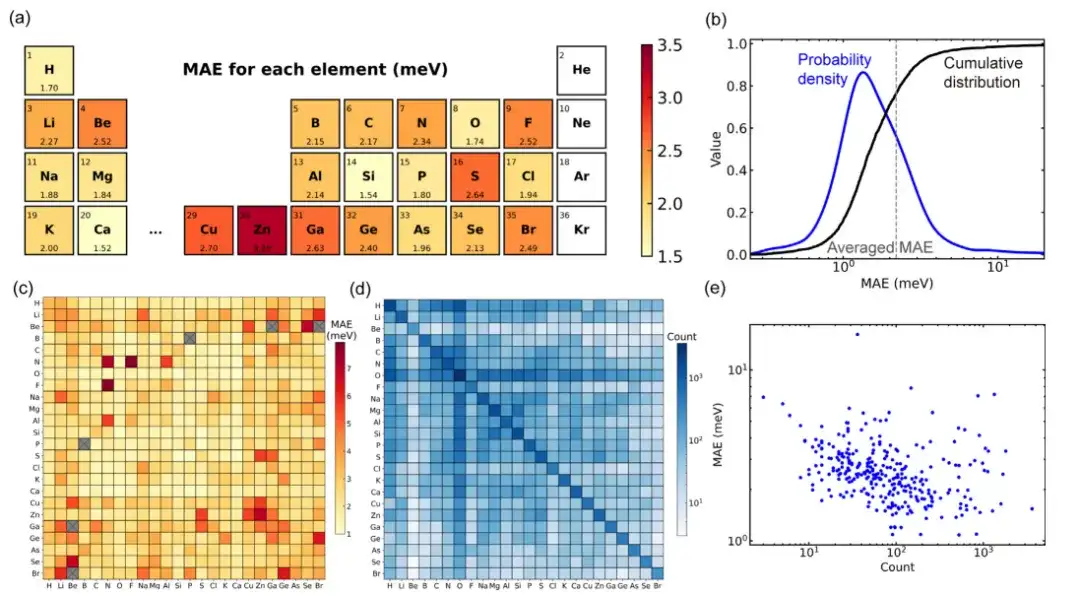
Bei der Bewertung der Leistung eines allgemeinen Materialmodells, das mit der Deep-2-Methode unter Verwendung einer großen Materialdatenbank mit 104 festen Materialien trainiert wurde, wiesen etwa 80 % der Materialstrukturen einen mittleren absoluten Fehler auf, der unter dem Mittelwert (2,2 meV) aller Strukturen im Datensatz lag. Nur 34 Strukturen (etwa 1,4 % des Testsatzes) weisen einen mittleren absoluten Fehler von mehr als 10 meV auf, was darauf hindeutet, dass das Modell eine gute Vorhersagegenauigkeit für Mainstream-Strukturen aufweist.
Durch eine weitere Analyse des Datensatzes kann die Abweichung in der Leistung des Modells hinsichtlich der Materialstruktur auf die Abweichung in der Datensatzverteilung zurückzuführen sein.Die Studie ergab, dass der entsprechende mittlere absolute Fehler umso kleiner ist, je mehr Trainingsstrukturen von Elementpaaren im Datensatz enthalten sind. Dieses Phänomen könnte darauf hinweisen, dass es bei allgemeinen Materialmodellen für Deep Learning ein „Skalierungsgesetz“ gibt, d. h. größere Trainingsdatensätze können die Modellleistung verbessern.
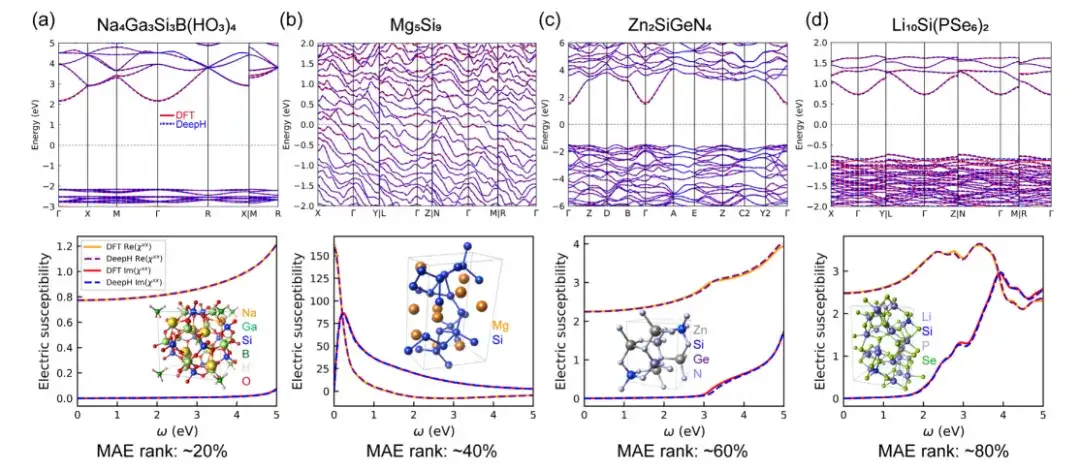
Um die Genauigkeit des allgemeinen DeepH-Materialmodells bei der Vorhersage von Materialeigenschaften zu bewerten, wurden in dieser Studie auf der Dichtefunktionaltheorie (DFT) basierende und von DeepH vorhergesagte DFT-Hamiltonoperatoren zur Berechnung von Beispielen verwendet und anschließend die mit diesen beiden Methoden erzielten Berechnungsergebnisse verglichen. Die Ergebnisse zeigen, dassDie von DeepH vorhergesagten Ergebnisse liegen sehr nahe an den von DFT berechneten Ergebnissen und zeigen die hervorragende Vorhersagegenauigkeit von DeepH bei der Berechnung von Materialeigenschaften.
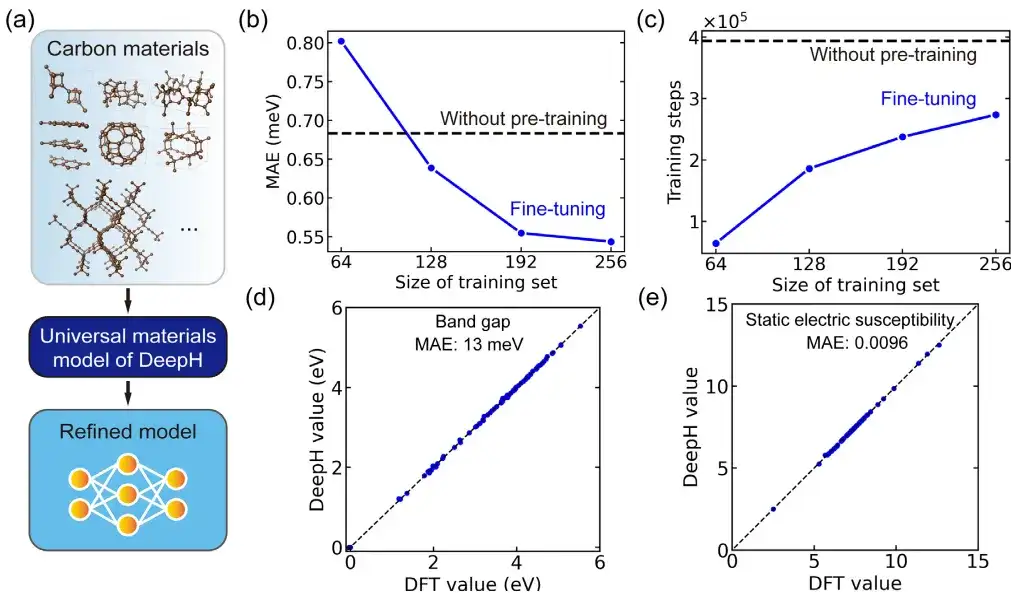
In einer spezifischen Anwendung untersuchte die Studie Kohlenstoffallotrope mithilfe eines fein abgestimmten allgemeinen Materialmodells. Darunter stammt der Kohlenstoffmaterialdatensatz aus der Samara Carbon Allotrope Database (SACADA), die insgesamt 427 Kohlenstoffallotrope mit unterschiedlichen Atomstrukturen enthält.
Darauf aufbauend haben die Forscher das allgemeine Materialmodell verfeinert und ein verbessertes DeepH-Modell speziell für Kohlenstoffmaterialien erstellt. Im Vergleich zum Modell ohne Vortraining kann durch die Feinabstimmung der mittlere absolute Fehler des vorhergesagten DFT-Hamiltonoperators deutlich auf 0,54 meV reduziert werden und auch bei Trainingsstrukturen mit weniger als 50% eine vergleichbare Vorhersagegenauigkeit erreicht werden.
Darüber hinaus verbessert die Feinabstimmung die Trainingskonvergenz erheblich und verkürzt die Trainingszeit. Man kann sagen, dass Feinabstimmung dazu beiträgt, die Vorhersagegenauigkeit zu verbessern und die Trainingseffizienz zu steigern. Und was noch wichtiger ist:Das fein abgestimmte DeepH-Modell zeigte erhebliche Vorteile bei der Vorhersage von Materialeigenschaften. Das fein abgestimmte Modell kann genaue Bandstrukturvorhersagen für fast alle getesteten Strukturen liefern.
Das Materialmodell wächst rasant, AI4S hat noch einen langen Weg vor sich
Mit ChatGPT als Ausgangspunkt ist die KI offiziell in eine neue „Ära der großen Modelle“ eingetreten. Diese Ära ist durch die Verwendung riesiger Datensätze und fortschrittlicher Algorithmen zum Trainieren von Deep-Learning-Modellen gekennzeichnet, die komplexe Aufgaben bewältigen können.Im Bereich der Materialwissenschaften läuten diese großen Modelle in Kombination mit der Weisheit der Forscher eine beispiellose neue Ära der Forschung ein.Diese großen Modelle können nicht nur riesige Mengen wissenschaftlicher Daten verarbeiten und analysieren, sondern auch die Eigenschaften und das Verhalten von Materialien vorhersagen. Dadurch beschleunigen sie die Entdeckung und Entwicklung neuer Materialien und lenken dieses Forschungsgebiet in eine effizientere und präzisere Richtung.
In der letzten Zeit kam es immer wieder zu Kollisionen zwischen der KI für die Wissenschaft und der Materialwissenschaft, wodurch neue Impulse entstanden.
Sitz in China,Die SF10-Gruppe des Nationalen Forschungszentrums für Festkörperphysik in Peking, das Institut für Physik der Chinesischen Akademie der Wissenschaften und das Computernetzwerk-Informationszentrum der Chinesischen Akademie der Wissenschaften arbeiteten zusammen, um Zehntausende von Daten zu chemischen Synthesepfaden in das große Modell LLAMA2-7b einzuspeisen und so das MatChat-Modell zu erhalten, mit dem der Synthesepfad anorganischer Materialien vorhergesagt werden kann. Die University of Electronic Science and Technology of China hat in Zusammenarbeit mit der Fudan University und dem Ningbo Institute of Materials Technology and Engineering der Chinese Academy of Sciences erfolgreich „ermüdungsbeständige ferroelektrische Materialien“ entwickelt und ist damit weltweit führend bei der Überwindung des Ermüdungsproblems ferroelektrischer Materialien, das die Branche seit mehr als 70 Jahren plagt. Das AIMS-Labor der Shanghai Jiao Tong University hat eine neue Generation des intelligenten Materialdesignmodells Alpha Mat entwickelt. … Es gibt immer wieder neue Forschungsergebnisse und eine neue Ära der Materialinnovation und -entdeckung ist angebrochen.
Wenn wir uns in der Welt umsehen,Googles DeepMind hat GNoME entwickelt, ein auf künstlicher Intelligenz basierendes Verstärkungslernmodell für die Materialwissenschaft, das mehr als 380.000 thermodynamisch stabile kristalline Materialien gefunden hat, was „800 Jahren intellektueller Ansammlung für die Menschheit“ entspricht und die Forschungsgeschwindigkeit bei der Entdeckung neuer Materialien erheblich beschleunigt; MatterGen von Microsoft, ein auf künstlicher Intelligenz basierendes Generierungsmodell im Bereich der Materialwissenschaften, kann auf Anfrage die Struktur neuer Materialien basierend auf den erforderlichen Materialeigenschaften vorhersagen. Meta AI hat mit amerikanischen Universitäten zusammengearbeitet, um den branchenweit führenden Datensatz für katalytische Materialien (Open Catalyst Project) und den Datensatz für die Adsorption organischer Metallgerüste (OpenDAC) zu entwickeln. Technologiegiganten haben mit ihren eigenen Technologien das Feld der Materialwissenschaften aufgemischt.
Im Vergleich zu herkömmlichen Methoden der Materialforschung und -entwicklung ermöglicht künstliche Intelligenz die Erforschung eines breiteren Spektrums an Materialmöglichkeiten und reduziert den mit der Materialentdeckung verbundenen Zeit- und Kostenaufwand erheblich. Allerdings steht „KI für die Wissenschaft“ im Bereich der Werkstoffe noch immer vor Herausforderungen hinsichtlich Glaubwürdigkeit und effektiver Umsetzung. Dazu gehören die Sicherstellung der Datenqualität sowie die Identifizierung und Minderung potenzieller Verzerrungen in den Daten, die zum Trainieren von KI-Systemen verwendet werden. Dies könnte auch bedeuten, dass es noch ein langer Weg ist, bis künstliche Intelligenz im Bereich der Materialwissenschaften eine größere Rolle spielen kann.








