Command Palette
Search for a command to run...
Veröffentlicht Im Cell Journal! Die Von Zhang Qiangfeng an Der Tsinghua-Universität Geleitete Forschungsgruppe Entwickelte Den SPACE-Algorithmus, Der Unter Ähnlichen Tools Über Die Führende Fähigkeit Zur Entdeckung Organisatorischer Module verfügt.
Obwohl Zellen in mehrzelligen Organismen dasselbe Genom besitzen, weisen sie aufgrund von Unterschieden in ihren internen Genregulationsnetzwerken und im externen Signalaustausch mit benachbarten Zellen in der umgebenden Mikroumgebung erhebliche Unterschiede in Morphologie, Genexpression und Funktion auf. Um Zelltypinformationen mit ihrer räumlichen Lage innerhalb von Geweben zu verknüpfen, wurde die Spatial Transcriptomics (ST)-Technologie entwickelt.Mit dieser Technologie können nicht nur hochauflösende Transkriptomdaten gewonnen werden, sondern auch entsprechende Positionsinformationen bereitgestellt werden, um die räumliche Verteilung und Positionsbeziehung verschiedener Zellsubtypen oder Transkriptionszustände zu bestimmen. Es spielt eine Schlüsselrolle beim Neuverständnis der Lebensstruktur, der individuellen Entwicklung, der Lebensentwicklung und der Definition von Krankheiten.
Dank der kontinuierlichen Weiterentwicklung der räumlichen Transkriptomik-Technologie sind Forscher in den letzten Jahren in der Lage, das Genexpressionsprofil von Zellen mit Einzelzellauflösung zu erhalten und gleichzeitig die räumlichen Positionsinformationen der Zellen innerhalb des Gewebes beizubehalten. Die effektive Nutzung dieser räumlichen Informationen zur Identifizierung räumlicher Zellsubtypen und zur Entdeckung von Gewebemodulen ist zu einer Kernaufgabe der räumlichen Transkriptomdatenanalyse geworden.
Derzeit ist die Analyse räumlicher Transkriptomdaten mit den folgenden zwei Herausforderungen konfrontiert: Erstens verwenden viele Studien zur Identifizierung räumlicher Zelltypen nur Zellgenexpressionsprofile und ignorieren die räumlichen Standortinformationen der Zellen. Forschungen der letzten Jahre haben gezeigt, dass Zelltypen, die ursprünglich als homogen galten, je nach ihrer Lage im Gewebe noch weiter in mehrere Untertypen unterteilt werden können. Zweitens ist es bei den Haargewebemodulen so, dass die Genexpressionsmerkmale der verschiedenen Zellen, aus denen das Gewebe besteht, sehr heterogen sein können. Daher ist es früheren Analysemethoden nicht gelungen, die Heterogenität der Zelltypen in räumlichen Transkriptomdaten mit Einzelzellauflösung voll auszunutzen.
Auf dieser GrundlageForschungsgruppe von Associate Professor Zhang Qiangfeng an der School of Life Sciences, Tsinghua University/Structural Biology Advanced Innovation Center/Tsinghua-Peking University Joint Center for Life Sciences,Eine Forschungsarbeit mit dem Titel „Tissue module discovery in single-cell resolution spatial transcriptomics data via cell-cell interaction-aware cell embedding“ (Entdeckung von Gewebemodulen in räumlichen Transkriptomikdaten mit Einzelzellauflösung über die Einbettung von Zellen zwischen Zellen) wurde kürzlich online in der Fachzeitschrift Cell Systems veröffentlicht.
Die Studie entwickelte einen künstlichen Intelligenzalgorithmus SPACE (räumliche Transkriptomik-Datenanalyse durch „interaktionsbewusste“ Zelleinbettung), der auf dem Graph-Autoencoder-Deep-Learning-Framework basiert.Die Fähigkeit, räumliche Zelltypen zu identifizieren und Gewebemodule aus räumlichen Transkriptomdaten mit Einzelzellauflösung zu entdecken, kann für groß angelegte räumliche Transkriptomstudien genutzt werden.
Forschungshighlights:
* Entwicklung von SPACE, einem KI-basierten Tool zur räumlichen Transkriptomanalyse, das räumliche Zelltypen identifizieren und Gewebemodule aus räumlichen Transkriptomdaten mit Einzelzellauflösung entdecken kann
* SPACE übertrifft andere Werkzeuge bei der Identifizierung von Zelltypen und der Entdeckung von Gewebemodulen deutlich, insbesondere bei komplexen Geweben mit mehreren Zelltypen
* SPACE kann für groß angelegte räumliche Transkriptomstudien verwendet werden, um zu verstehen, wie Interaktionen zwischen räumlich benachbarten Zellen die biologischen Funktionen von Zelltypen und Gewebemodulen beeinflussen
Papieradresse:
https://www.cell.com/cell-systems/fulltext/S2405-4712(24)00124-8
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensätze: Mehrere Datensätze überprüfen die SPACE-Funktionen
Um die Fähigkeiten von SPACE zu überprüfen, wurden in der Studie mehrere Datensätze verwendet, die wie folgt zusammengefasst werden:
Adresse zum Herunterladen des Datensatzes:
https://go.hyper.ai/CBJfX
MERFISH-Maus-PMC-Datensatz
Für den MERFISH-Maus-PMC-Datensatz wurde die logarithmisch transformierte normalisierte Zell-Gen-Matrix aus der Brain Image Library bezogen und Zellen, die als „andere“ gekennzeichnet waren oder sich außerhalb der Hauptprobenregion befanden, wurden entfernt.
Datensatzlink:
STARmap Maus-PLA-Datensatz
Für den STARmap-Maus-PLA-Datensatz wurde die normalisierte Zell-Gen-Matrix durch das Originalpapier bereitgestellt und log-transformiert.
Datensatzlink:
https://drive.google.com/file/d/1DDCowUuZ7PPFUSZsjvSqntWkYJMjf1Na/view?usp=sharing
MERFISH Maus AB Datensatz
Für den MERFISH-Maus-AB-Datensatz wurde die Genzählmatrix aus der CELL x GENE-Bibliothek bezogen. Die Gesamtzahlen pro Zelle wurden auf 10.000 normalisiert und die normalisierte Zell-Gen-Matrix wurde log-transformiert.
Datensatzlink:
https://cellxgene.cziscience.com/collections/31937775-06024e52-a799-b6acdd2ba2e
MERFISH Maus-WB-Datensatz
Für den MERFISH-Maus-WB-Datensatz wurde die logarithmisch transformierte normalisierte Zell-Gen-Matrix aus dem GitHub-Repository bezogen.
Datensatzlink:
https://github.com/AllenInstitute/abc_atlas_access
Xenium Human BC-Datensatz
Für den Xenium-Human-BC-Datensatz wurde die Genzählmatrix von der 10x-Genomics-Website bezogen. Die Gesamtzahlen pro Zelle wurden auf 10.000 normalisiert und die normalisierte Zell-Gen-Matrix wurde log-transformiert.
Datensatzlink:
https://www.10xgenomics.com/products/xenium-in-situ/preview-dataset-human-breast
CosMx Human NSCLC-Datensatz
Für den CosMx-Datensatz zum menschlichen NSCLC wurde die logarithmisch transformierte normalisierte Zell-Gen-Matrix von der nanoString-Website bezogen.
Datensatzlink:
https://nanostring.com/products/cosmx-spatial-molecular-imager/ffpe-dataset/nsclc-ffpe-dataset
Visium-Datensatz zum menschlichen Gehirn
Für den Visium-Datensatz zum menschlichen Gehirn wurde die Genzählmatrix mithilfe des Bioconductor-Pakets spatialLIBD ermittelt. Die 3.000 am stärksten variablen Gene wurden in jeder Probe des Visium-Datensatzes zum menschlichen Gehirn mithilfe der Funktion scanpy.pp.highly_variable_genes() (Flavor = „seurat_v3“) des Python-Pakets SCANPY (v1.9.1) identifiziert. Die Gesamtzahlen pro Zelle wurden dann auf 10.000 normalisiert und die normalisierte Zell-Gen-Matrix wurde log-transformiert.
Datensatzlink:
https://bioconductor.org/packages/release/data/experiment/html/spatialLIBD.html
Modellarchitektur: Ein zellinteraktionsbewusstes, zelleneingebettetes Modell
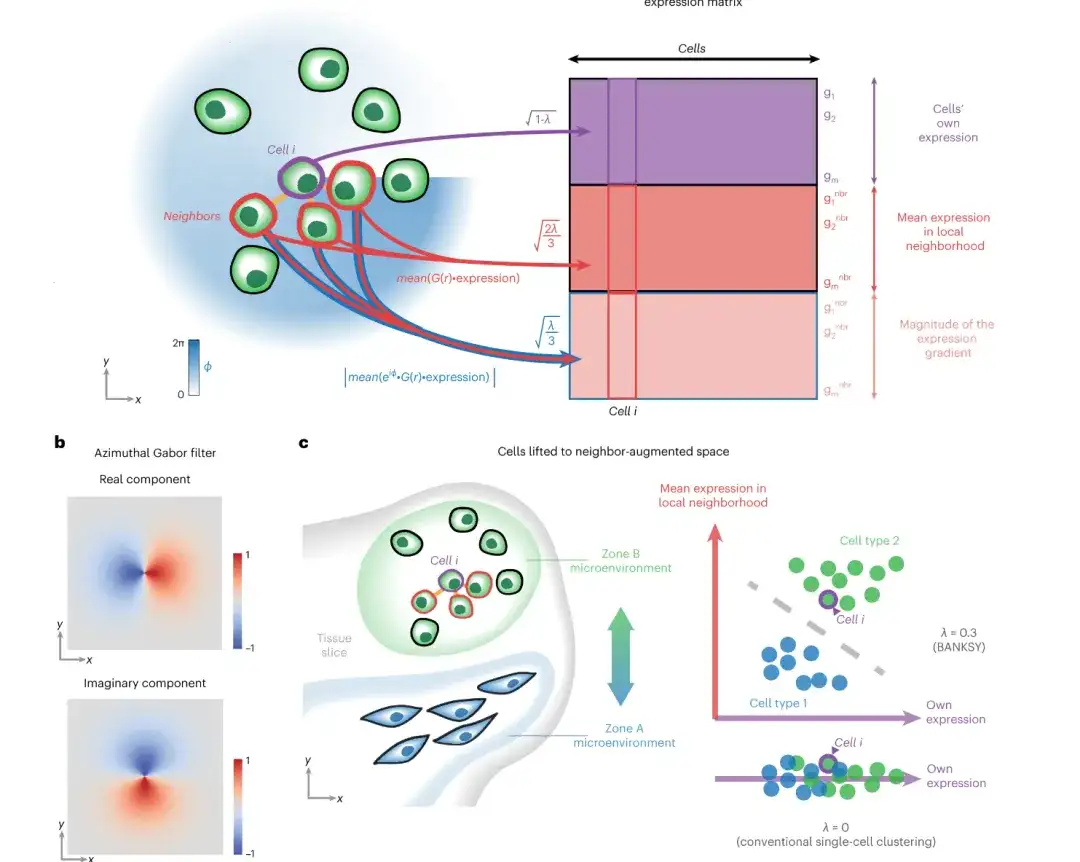
SPACE verwendet ein Graph-Autoencoder-Framework, um niedrigdimensionale Zelleinbettungen zu erlernen, die die Genexpressionsdaten jeder Zelle in den räumlichen Transkriptomdaten sowie deren Interaktionsdaten mit räumlich benachbarten Zellen beschreiben (daher wird die Zelleinbettung als „Zell-Zell-Interaktions-bewusste Zelleinbettung“ bezeichnet). Basierend auf dieser Zelleinbettung verwendet SPACE dann Clustering-Algorithmen, um räumliche Zellsubtypen zu identifizieren und Gewebemodule zu entdecken.
Aus der Perspektive der ArchitekturDas SPACE-Modell besteht aus drei Teilen: Encoder (dreischichtiges Graph-Attention-Netzwerk), Nachbargraph-Decoder und Genexpression-Decoder.Die folgende Abbildung zeigt den Gesamtrahmen des Modells:
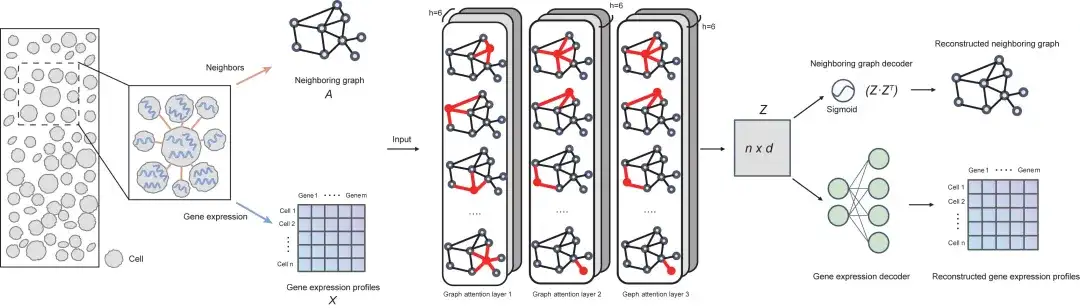
Zunächst erstellt SPACE einen Adjazenzgraphen, indem es jede Zelle auf der Grundlage der räumlichen Nähe mit ihren k nächsten Nachbarzellen verbindet. Anschließend verwendet SPACE ein dreischichtiges Graph Attention Network (GAT) als Encoder, um die Eingabe des Genexpressionsprofils und des Adjazenzgraphen in niedrigdimensionale Zelldarstellungen umzuwandeln, die anschließend verwendet werden, um das Genexpressionsprofil und den Adjazenzgraphen jeder Zelle durch zwei unabhängige Decodernetzwerke zu rekonstruieren.
Zum Trainieren des GAE-Modells verwendet SPACE selbstüberwachtes Lernen mit dem Ziel, den Gesamtverlust bei der Rekonstruktion von Genexpressionsprofilen und Adjazenzgraphen zu minimieren. Die erlernten Zelldarstellungen können dann mithilfe verschiedener Clustering-Algorithmen zur Zelltypidentifizierung und zur Erkennung organisatorischer Module verwendet werden.
Zuvor entwickelte Deep-Learning-Tools haben Graph-Convolutional-Netzwerke (GCNs) (z. B. SpaGCN, SpaceFlow, GraphST und SEDR) oder Graph-Attention-Autoencoder (z. B. STAGATE) verwendet, um „nachbarschaftsbewusste“ Einbettungen zu generieren, die Organisationsmodule entdecken, indem sie die Genexpressionsprofile von Zellen und ihren Nachbarn aggregieren. SPACE unterscheidet sich von diesen Tools in drei wesentlichen Punkten:
Zunächst muss SPACE sowohl Genexpressionsprofile als auch Adjazenzgraphen aus derselben niedrigdimensionalen Zelldarstellung rekonstruieren (über zwei unabhängige Decoder).Dieses Design ermöglicht es SPACE, sich die Genexpressionsprofile und räumlichen Interaktionen der analysierten Zelle und ihrer einzelnen Nachbarzellen zu merken. Im Gegensatz dazu verwenden andere Methoden den Adjazenzgraphen als Eingabe, rekonstruieren den Graphen jedoch nicht. Um diesen Unterschied hervorzuheben, werden die von SPACE generierten Zelleinbettungen in dieser Studie als „Zell-Zell-Interaktions-bewusste Zelleinbettungen“ bezeichnet.
Zweitens definiert SPACE ein rezeptives Domänenverhältnis, um die relativen Gewichte der Verluste des Genexpressionsprofils und der Rekonstruktion des Adjazenzgraphen zu bestimmen.Dieses anpassbare Verhältnis ermöglicht es SPACE, den Lernschwerpunkt an spezifische Forschungsanforderungen anzupassen und das Genexpressionsprofil jeder analysierten Zelle oder die Interaktionen räumlich benachbarter Zellen hervorzuheben.
Drittens verwendet SPACE auch den Aufmerksamkeitsmechanismus im GAT-Encoder, um während des Prozesses der Nachbarschaftsinformationsaggregation adaptiv die Gewichtung jeder Nachbarschaft zu erlernen.Dieser Ansatz berücksichtigt automatisch die jeweiligen Beiträge verschiedener Nachbarschaften bei der Rekonstruktion von Genexpressionsprofilen.
Forschungsergebnisse: SPACE übertrifft andere ähnliche Tools bei der Identifizierung von Zelltypen und der Entdeckung von Gewebemodulen
SPACE wurde anhand mehrerer räumlicher Transkriptom-Datensätze getestet. Dabei zeigte sich, dass die von SPACE entdeckten Zellgemeinschaften in ihren räumlichen Verteilungsmerkmalen manuell annotierten Gewebestrukturen ähnelten.
Bewertung der Fähigkeit von SPACE, räumlich informative Zelltypen zu identifizieren
Wir verwendeten zunächst einen mit MERFISH charakterisierten ST-Datensatz des primären Motorkortex (PMC) der Maus (beginnend mit Schnitt 153), um die Fähigkeit von SPACE zur Identifizierung von Zelltypen zu untersuchen. Die Ergebnisse zeigen, dassDie von SPACE identifizierten Zelltypen stimmten gut mit denen überein, die in der ursprünglichen Studie berichtet wurden.Wie in der Abbildung unten gezeigt; Darüber hinaus bietet SPACE auch Zelltypanmerkungen mit höherer Auflösung für bestimmte Zelltypen (wie Astrozyten und Oligodendrozyten).
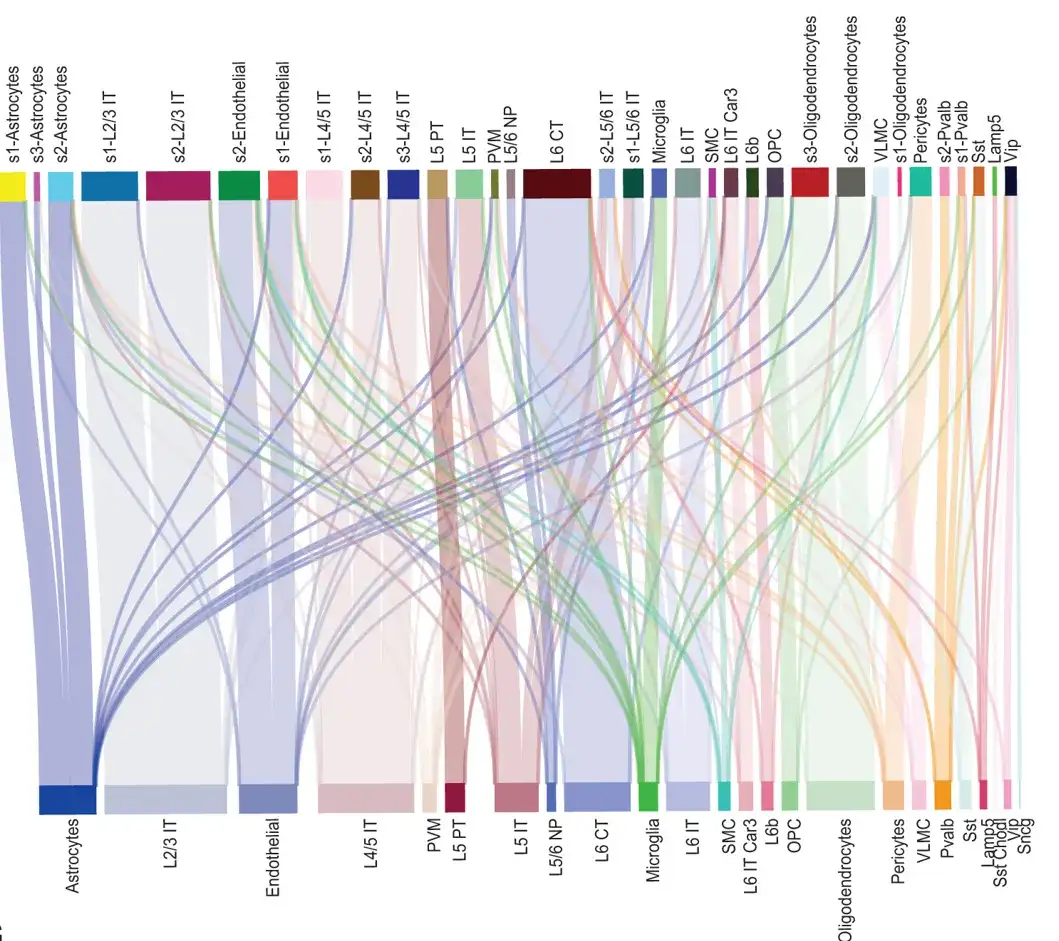
Sankey-Diagramm, das die Entsprechung zwischen den relevanten Zelltypen und den ursprünglichen Zelltypen in den räumlichen Informationen aller Zellen in Abschnitt 153 des MERFISH-Maus-PMC-Datensatzes zeigt
Anschließend konzentrierten sich die Forscher weiter auf die identifizierten Subtypen von Astrozyten (Gliazellen im Kortex) und Oligodendrozyten (myelinisierende Zellen im zentralen Nervensystem). Astrozyten galten einst als homogener Zelltyp, neuere ST-Studien zeigen jedoch, dass sie in unterschiedlichen Gehirnregionen unterschiedliche Funktionen haben.
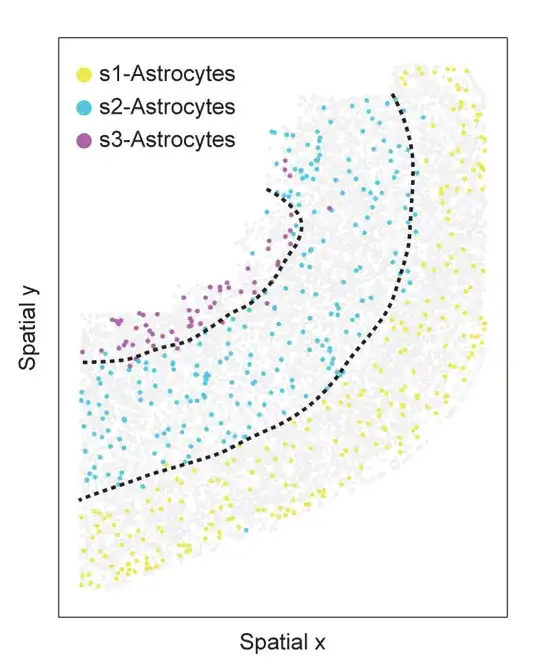
Räumliche informationsbezogene Astrozyten-Subtypen in Schnitt 153 des MERFISH-Maus-PMC-Datensatzes. Die Zellen sind nach Astrozyten-Subtyp gefärbt und hellgraue Punkte stellen andere Zellen dar. Gestrichelte Linien zeigen obere, tiefe und weiße Substanz an
Im Experiment fand SPACE drei verschiedene Subtypen im PMC-Schnitt 153, wie in der Abbildung oben gezeigt, wobei jeder Subtyp räumlich in einer anderen kortikalen Schicht verteilt war. Ähnlich wie Astrozyten klassifiziert SPACE auch Oligodendrozyten in drei räumlich informative Subtypen mit unterschiedlichen räumlichen Verteilungsmustern.

Die Forscher wendeten SPACE auch auf einen Datensatz zur Mausplazenta (PLA) an, der mit STARmap, einer anderen ST-Technologie, generiert wurde. Die Ergebnisse zeigen, dass SPACE die Zellen in 16 Zelltypen einteilte, die, wie oben gezeigt, gut mit den Zelltypen in der ursprünglichen Studie übereinstimmten. SPACE identifizierte zwei Glykotrophoblasten-Subtypen, die in der ursprünglichen Studie beide als „Megatrophoblasten 2“-Zellen bezeichnet wurden. Die beiden Subtypen sind in unterschiedlichen Regionen der Plazenta lokalisiert und haben unterschiedliche benachbarte, interagierende Zelltypen.
Zusammenfassend lässt sich sagen, dass die Analyse zweier unabhängiger Datensätze, die auf unterschiedlichen ST-Methoden und -Organisationen basieren, die folgenden Schlussfolgerungen stützt:SPACE kann biologisch unterschiedliche Zelltypen anhand der räumlichen Informationen im ST-Datensatz identifizieren.
Bewertung der Leistung von SPACE bei der Zelltypidentifizierung
Die Forscher verglichen SPACE mit zwei aktuellen Tools zur Identifizierung von Zelltypen aus räumlichen Transkriptomikdaten, BANKSY und FICT, die neben der Genexpression auch räumliche Informationen berücksichtigen. In ihre Analyse schlossen die Forscher auch SCANPY ein, ein weit verbreitetes Tool zur Zelltypidentifizierung, das allerdings nur die Genexpression berücksichtigt.
Zum Vergleich verwendeten die Forscher den zuvor erwähnten MERFISH-Maus-PMC-Datensatz und den STARmap-Maus-PLA-Datensatz. Wie in der Abbildung unten gezeigt, kann SPACE verschiedene räumlich informierte Astrozyten- und Oligodendrozyten-Subtypen identifizieren, aber weder SCANPY noch FICT können Astrozyten- und Oligodendrozyten-Subtypen mit räumlichen Verteilungsmustern aufgelöst in den kortikalen Schichten definieren.
Obwohl SPACE und BANKSY im STARmap-Maus-PLA-Datensatz die beiden Glykotrophoblasten-Subtypen erfolgreich identifizierten, gelang es SCANPY und FICT nicht, die Glykotrophoblasten-Subtypen zu identifizieren. Dies kann an den offensichtlichen Unterschieden in den umgebenden Zelltypen zwischen den beiden Glykotrophoblasten-Subtypen liegen.
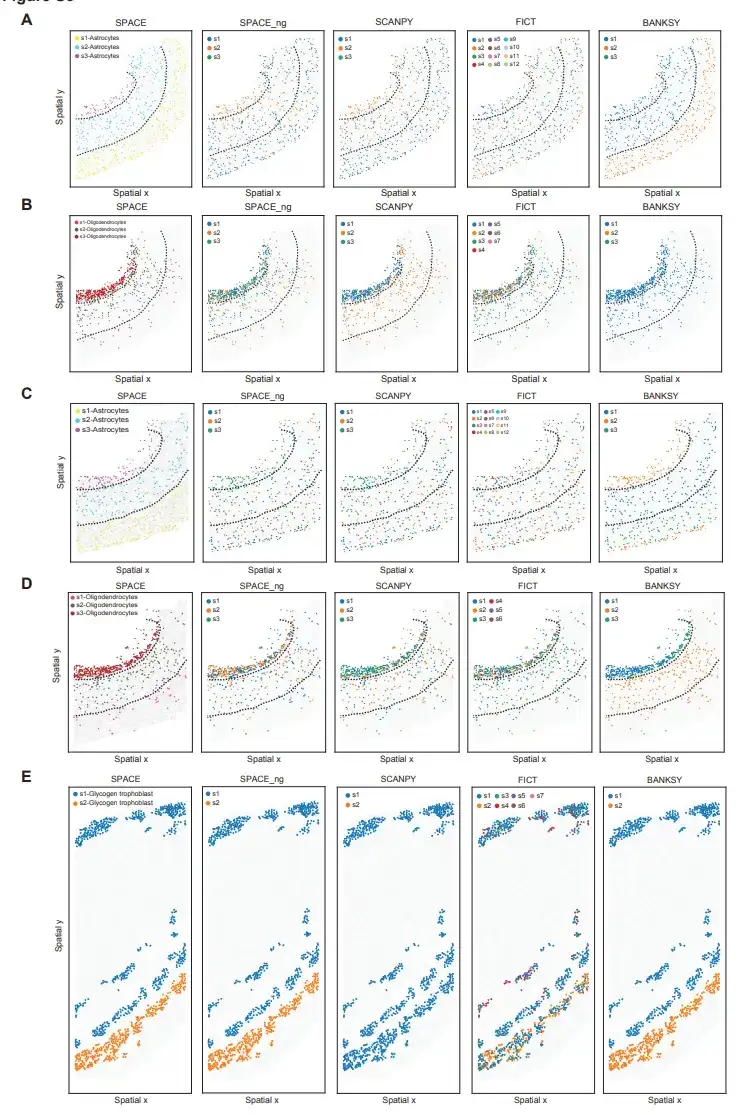
Diese Ergebnisse zeigen insgesamt, dassSPACE übertrifft derzeit verfügbare Tools bei der Unterscheidung räumlich informativer Zelltypen von ST-Daten.
SPACE übertrifft modernste Werkzeuge bei der Entdeckung von Gewebemodulen
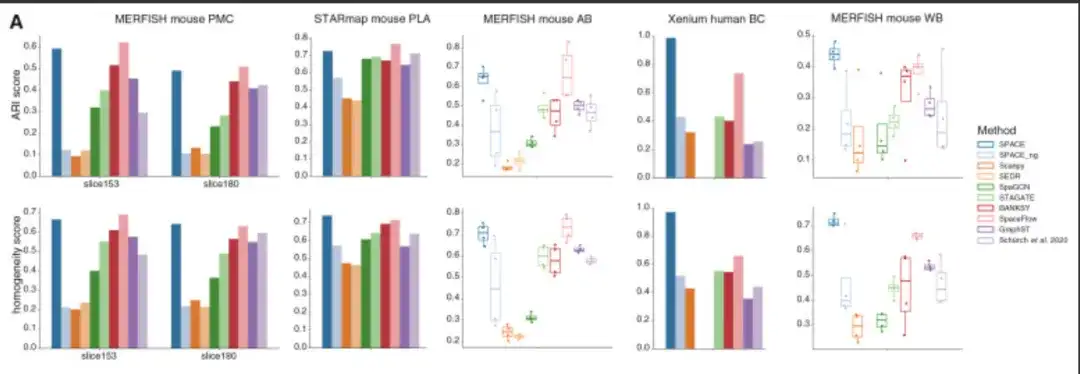
Eine wichtige Aufgabe in räumlichen Transkriptomikstudien besteht darin, Organisationsmodule innerhalb eines bestimmten Gewebes zu entdecken. Um die Fähigkeiten von SPACE in dieser Hinsicht zu bewerten, verglichen die Forscher SPACE mit SEDR, SpaGCN, STAGATE, BANKSY, SpaceFlow, GraphST, den Methoden von Schürch et al. sowie SCANPY und SPACE_ng und verwendeten zwei der oben genannten ST-Datensätze (MERFISH-Maus-PMC-Datensatz und STARmap-Maus-PLA-Datensatz) sowie drei zusätzliche Datensätze mit annotierten Gewebemodulen, darunter den MERFISH-Datensatz zum gealterten Maushirn (AB), den MERFISH-Datensatz zum gesamten Maushirn (WB) und den Xenium-Datensatz zum menschlichen Brustkrebs (BC), die ST-Daten darstellen, die aus verschiedenen Geweben und unter verschiedenen Bedingungen gewonnen wurden.
Gesamt,SPACE übertrifft die Konkurrenztools in 2 der 5 Datensätze bei weitem und weist in den anderen 3 Datensätzen eine fast ebenso gute Leistung auf wie die Tools mit der besten Leistung (im Verhältnis zu den jeweils besten Tools).Wie in der folgenden Abbildung dargestellt:
Bewältigung der Herausforderungen der räumlichen Transkriptomdatenanalyse
Die Spatial Transcriptomics-Technologie ist einer der größten Durchbrüche auf dem Gebiet der Bioinformatik der letzten Jahre und wurde 2020 von Nature Method zur Technologie des Jahres gekürt.Diese Technologie gleicht den Mangel aus, der darin besteht, dass die Einzelzellsequenzierungstechnologie Schwierigkeiten bei der Messung der Positionsbeziehungen zwischen einzelnen Zellen hat, indem sie gleichzeitig die räumliche Position einer großen Anzahl von Zellen und die Transkriptomzählungen innerhalb der Zellen misst und so eine neue Datengrundlage für das Verständnis der Interaktionen zwischen mehreren Zellen bereitstellt. Die Entwicklung grundlegender Analysemethoden für räumliche Transkriptomdaten ist eines der aktuellen Spitzenthemen im Bereich der Bioinformatik.
Durch die Kopplung von Informationen zur räumlichen Lokalisierung von Zellen und ihrem molekularen Eigenschaftsspektrum ist eine neue Art multimodaler Hochdurchsatz-Datenressource entstanden, die viele Herausforderungen an die Entwicklung effizienter Methoden zur Datenanalyse und zum Information Mining stellt. Künstliche Intelligenz liefert neue Ideen zur Lösung dieser Herausforderungen.

Im Juli 2022 veröffentlichte die Forschungsgruppe von Professor Shen Hongbin und Associate Professor Yuan Ye von der Abteilung für Automatisierung der Fakultät für elektronische Informations- und Elektrotechnik der Shanghai Jiao Tong University eine Forschungsarbeit mit dem Titel „Zellclustering für räumliche Transkriptomikdaten mit Graph-Neuralnetzwerken“ in Nature Computational Science, einer Tochtergesellschaft von Nature.
Link zum Artikel:https://www.nature.com/articles/s43588-022-00266-5
Das Papier schlägt eine Methode zur räumlichen Transkriptom-Zellclusterung (Cell Clustering for Spatial Transcriptomics, CCST) auf der Grundlage eines Graph Convolutional Neural Network vor.Es bietet eine neue Lösung für die Verarbeitung räumlicher Transkriptomdaten und kann möglicherweise bei der Erforschung mehrstufiger grundlegender Probleme in den Biowissenschaften und der Medizin eingesetzt werden, darunter bei der Modellierung der räumlichen Verteilung der Genexpression, der Analyse der Zelldynamik und der Entdeckung wichtiger Interaktionen zwischen Zellsubtypen und ihrer molekularen Mechanismen.
April 2023Ein Forschungsteam der Johns Hopkins University hat SpaceMarkers entwickelt.Dabei handelt es sich um einen bioinformatischen Algorithmus, der mithilfe der latenten Raumanalyse von ST-Daten Rückschlüsse auf molekulare Veränderungen in Zell-Zell-Interaktionen ziehen kann. Mit diesem Ansatz konnten die Forscher anhand räumlicher Transkriptomikdaten von Visium Rückschlüsse auf molekulare Veränderungen der Tumor-Immun-Interaktionen bei metastasierten, invasiven und Vorläuferläsionen sowie als Reaktion auf eine Immuntherapie ziehen.
Die Studie wurde in Cell Systems unter dem Titel „Uncovering the spatial landscape of molecular interactions within the tumor microenvironment through latente spaces“ veröffentlicht.
Im April dieses Jahres wurde in der internationalen Fachzeitschrift Nature Genetics ein Forschungsbericht mit dem Titel „BANKSY vereinheitlicht Zelltypisierung und Gewebedomänensegmentierung für eine skalierbare räumliche Omics-Datenanalyse“ veröffentlicht.Wissenschaftler des A*STAR Research Institute in Singapur und anderer Institutionen berichteten über einen Algorithmus namens BBANKSY (Building Aggregates with a Neighborhood Kernel and Spatial Yardstick).Als innovatives Tool zur räumlichen Omics-Datenanalyse besteht die Hauptfunktion dieses Algorithmus darin, Zellen in räumlichen Omics-Daten effektiv nach Typ und Gewebedomäne zu klassifizieren.
Link zum Artikel:https://www.nature.com/articles/s41588-024-01664-3
Offensichtlich wird die räumliche Transkriptomik-Technologie mit Unterstützung der künstlichen Intelligenz in Zukunft die räumliche Verteilung verschiedener Zelltypen in Geweben sowie die Wechselwirkungen zwischen verschiedenen Zellpopulationen besser aufdecken und Genexpressionskarten verschiedener Geweberegionen erstellen können, was einen weitreichenden Anwendungswert für das Verständnis der Entstehungsmechanismen von Krankheiten und Krebs hat.
Quellen:
1.https://www.cell.com/cell-systems/fulltext/S2405-4712(24)00124-8#secsectitle0030
2.https://www.tsinghua.edu.cn/info/1175/112190.htm
3.https://news.bioon.com/article/367a820e60b9.html
4.https://www.sohu.com/a/677912398_12








