Command Palette
Search for a command to run...
Tsinghua AIR Und Andere Wurden Für ICML Ausgewählt Und Veröffentlichten Gemeinsam Das Proteinsprachenmodell ESM-AA, Das Das Traditionelle SOTA Übertrifft

Als treibende Kraft unzähliger biochemischer Reaktionen innerhalb von Zellen spielen Proteine die Rolle von Architekten und Ingenieuren in der mikroskopischen Welt der Zellen. Sie katalysieren nicht nur Lebensaktivitäten, sondern dienen auch als Grundkomponenten für den Aufbau und die Aufrechterhaltung der Morphologie und Funktion von Organismen. Es ist die Interaktion und Synergie zwischen Proteinen, die den großen Bauplan des Lebens unterstützen.
Allerdings ist die Struktur von Proteinen komplex und veränderlich und die Analyse der Proteinstruktur mit herkömmlichen experimentellen Methoden ist zeitaufwändig und mühsam. Es entstanden Proteinsprachmodelle (PLMs). Durch den Einsatz von Deep-Learning-Technologie und die Analyse einer großen Menge an Proteinsequenzdaten sowie das Erlernen der biochemischen Gesetze und Koevolutionsmuster von Proteinen wurden bemerkenswerte Erfolge in den Bereichen der Proteinstrukturvorhersage, der Anpassungsfähigkeitsvorhersage und des Proteindesigns erzielt und die Entwicklung des Protein-Engineerings erheblich vorangetrieben.
Obwohl PLMs auf der Rückstandsskala große Erfolge erzielt haben, sind sie in ihrer Fähigkeit, Informationen auf atomarer Ebene bereitzustellen, begrenzt. Als Reaktion darauf hat sich Zhou Hao, ein assoziierter Forscher am Institut für Intelligente Industrien der Tsinghua-Universität, mit der Peking-Universität, der Universität Nanjing und dem Shuimu Molecular Team zusammengetan, umEs wird ein mehrskaliges Proteinsprachenmodell ESM-AA (ESM All Atom) vorgeschlagen.Durch die Entwicklung von Trainingsmechanismen wie Residuenerweiterung und mehrskaliger Positionskodierung wurde die Fähigkeit zur Verarbeitung von Informationen im atomaren Maßstab erweitert.
Die Leistung von ESM-AA bei Aufgaben wie der Ziel-Liganden-Bindung wurde deutlich verbessert und übertrifft aktuelle SOTA-Proteinsprachenmodelle wie ESM-2 und auch aktuelle SOTA-Lernmodelle für molekulare Repräsentationen wie Uni-Mol. Verwandte Forschungsergebnisse wurden unter dem Titel „ESM All-Atom: Multi-scale Protein Language Model for Unified Molecular Modeling“ veröffentlicht.Veröffentlicht auf ICML, der führenden Konferenz zum maschinellen Lernen.

Papieradresse:
https://icml.cc/virtual/2024/poster/35119
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensatz: Es wurde ein gemischter Datensatz aus Protein- und Moleküldaten erstellt
In der VortrainingsaufgabeFür die Studie wurde ein kombinierter Datensatz aus Protein- und Moleküldaten verwendet, der Strukturinformationen wie etwa Atomkoordinaten enthielt.
Für den Proteindatensatz verwendete die Studie AlphaFold DB, das 8 Millionen mit hoher Zuverlässigkeit von AlphaFold2 vorhergesagte Proteinsequenzen und -strukturen enthält.
Für den molekularen Datensatz verwendete die Studie Daten, die von den molekularen Kraftfeldern ETKDG und MMFF generiert wurden und 19 Millionen Moleküle und 209 Millionen Konfigurationen enthalten.
Beim Training von ESM-AA mischten die Forscher zunächst einen Proteindatensatz Dp und einen Moleküldatensatz Dm zum endgültigen Datensatz zusammen, d. h. D=Dp∪Dm. Da Moleküle aus Dm nur aus Atomen bestehen, ist ihre Codekonvertierungssequenz X̄ die geordnete Menge aller Atome Ā ohne jegliche Reste, d. h. R̄=∅. Es ist erwähnenswert, dass ESM-AA sowohl Proteine als auch Moleküle als Eingabe akzeptieren kann, da beim Vortraining molekulare Daten verwendet werden.
ESM-AA-Modellkonstruktion: Multiskaliges Vortraining und Kodierung zur Erzielung einer einheitlichen molekularen Modellierung
Inspiriert von der Methode des mehrsprachigen Code-Switchings dekomprimiert ESM-AA bei der Durchführung von Vorhersage- und Proteindesignaufgaben zunächst einige Reste zufällig, um mehrskalige Code-Switching-Proteinsequenzen zu generieren. Diese Sequenzen werden dann durch sorgfältig entwickelte mehrskalige Positionskodierungen trainiert und ihre Wirksamkeit wurde auf der Rest- und Atomskala nachgewiesen.
Bei der Bearbeitung proteinmolekularer Aufgaben, also Aufgaben mit Proteinen und kleinen Molekülen, benötigt ESM-AA keine zusätzliche Modellunterstützung und kann die Fähigkeiten des vortrainierten Modells voll ausnutzen.
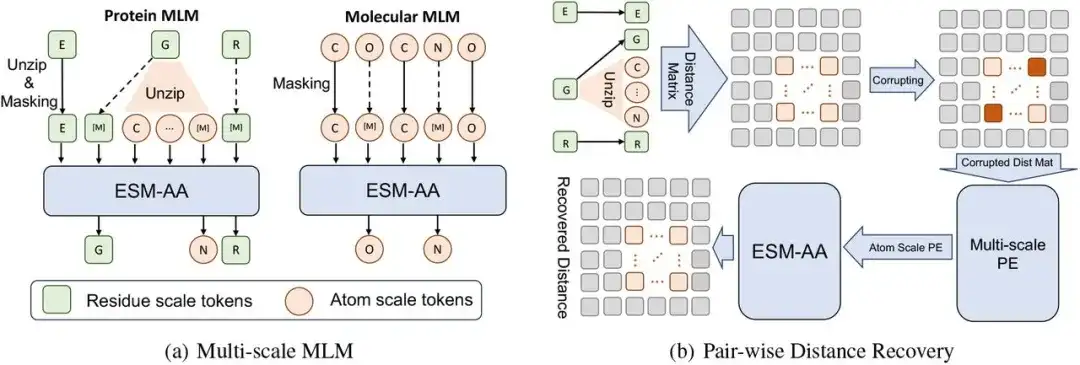
Das mehrskalige Vortrainingsframework dieser Studie besteht aus einem mehrskaligen maskierten Sprachmodell (MLM) und einer paarweisen Distanzwiederherstellung.
Insbesondere kann ein Protein X auf der Restskala als eine Sequenz von L Resten betrachtet werden, d. h. X = (r1, …, ri, …, rL). Jeder Rest ri besteht aus N Atomen A Ai={a1i,…,aNi}. Um die Code-Switching-Proteinsequenz X̅ zu konstruieren, haben wir einen Dekomprimierungsprozess implementiert, indem wir zufällig eine Reihe von Resten ausgewählt und ihre entsprechenden Atome in X eingefügt haben. In diesem Prozess haben die Forscher die dekomprimierten Atome der Reihe nach angeordnet und schließlich, nachdem sie den Atomsatz Ai in X eingefügt (d. h. den Rest ri dekomprimiert) hatten, eine Code-Switching-Sequenz X̄ erhalten.
Dann,Die Forscher führten eine maskierte Sprachmodellierung der Code-Switching-Sequenz X̄ durch.
Zuerst maskieren wir zufällig einige Atome oder Reste in X̄ und lassen das Modell die ursprünglichen Atome oder Reste anhand des umgebenden Kontexts vorhersagen. Die Forscher verwendeten dann Pairwise Distance Recovery (PDR) als weitere Vortrainingsaufgabe. Das heißt, die Strukturinformationen auf atomarer Ebene werden durch das Hinzufügen von Rauschen zu den Koordinaten zerstört und die zerstörten Informationen zum interatomaren Abstand werden als Modelleingabe verwendet, wodurch das Modell den genauen euklidischen Abstand zwischen diesen Atomen wiederherstellen muss.
Unter Berücksichtigung des semantischen Unterschieds zwischen den Strukturinformationen über große Entfernungen hinweg über verschiedene Reste hinweg und den Strukturinformationen auf atomarer Ebene innerhalb eines einzelnen Rests berechnet diese Studie nur den PDR innerhalb des Rests, was es ESM-AA auch ermöglichen kann, verschiedene Strukturkenntnisse innerhalb verschiedener Reste zu erlernen.
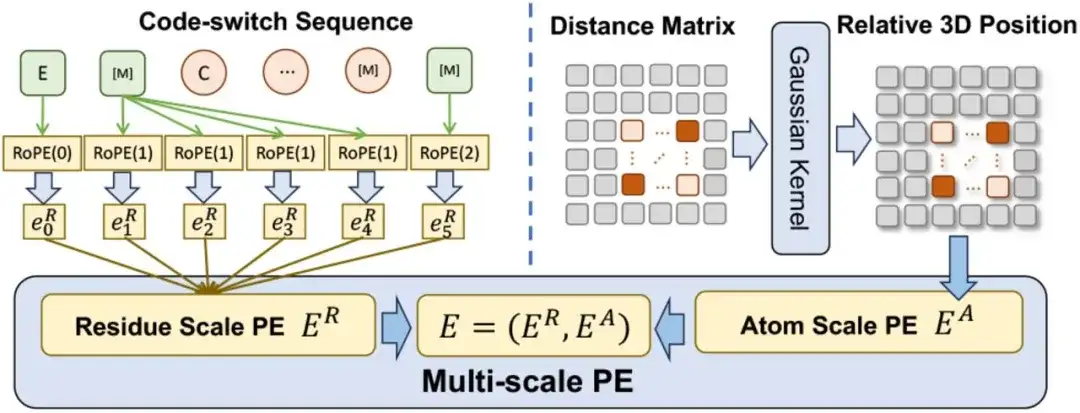
Im Hinblick auf die mehrskalige Positionskodierung haben die Forscher eine mehrskalige Positionskodierung E entwickelt, um die Positionsbeziehung in der Code-Switching-Sequenz zu kodieren. E enthält einen Positionscode ER auf der Restskala und einen Positionscode EA auf der Atomskala.
Für ER,Die Forscher erweiterten eine bestehende Kodierungsmethode, um die Kodierung von Rest-Atom-Beziehungen zu ermöglichen und gleichzeitig bei der Verarbeitung reiner Restsequenzen die Konsistenz mit der ursprünglichen Kodierung aufrechtzuerhalten.Für EA,Um die Beziehungen zwischen Atomen zu erfassen, verwendet die Studie direkt eine räumliche Distanzmatrix, um ihre dreidimensionalen Positionen zu kodieren.
Es ist erwähnenswert, dass die Multiskalen-Kodierungsmethode sicherstellt, dass das Vortraining nicht durch die mehrdeutige Positionsbeziehung beeinträchtigt wird, sodass ESM-AA auf beiden Skalen effektiv arbeiten kann.
Bei der Integration von PE mit mehreren Skalen in Transformer wurde in der Studie zunächst die Sinuscodierung in Transformer durch die Positionscodierung der Restskala ER ersetzt und die Positionscodierung der Atomskala EA als Bias-Term der Selbstaufmerksamkeitsschicht betrachtet.
Forschungsergebnisse: Integration molekularen Wissens zur Optimierung des Proteinverständnisses
Um die Wirksamkeit des vereinheitlichten, vortrainierten Modells mit mehreren Maßstäben zu überprüfen, wurde in dieser Studie die Leistung von ESM-AA bei verschiedenen Aufgaben mit Proteinen und kleinen Molekülen bewertet.
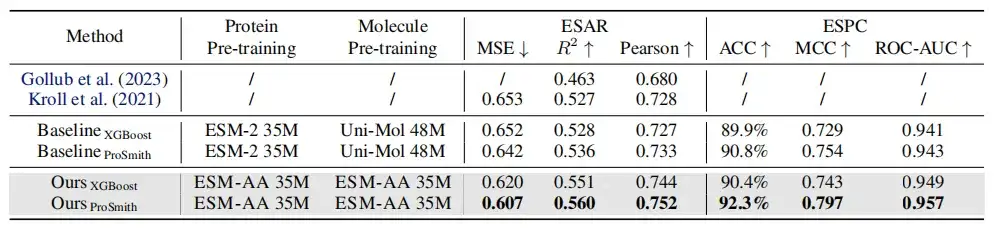
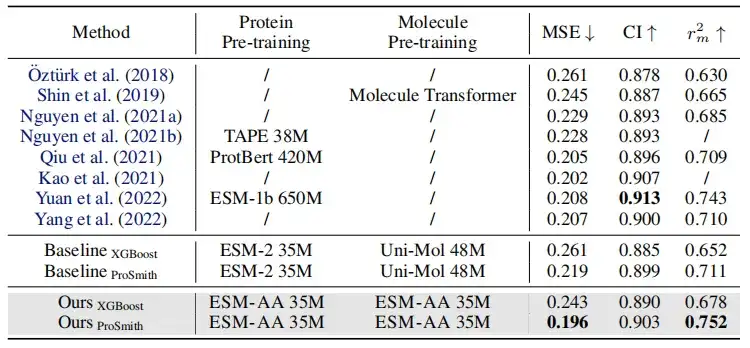
Wie in der obigen Tabelle gezeigt, zeigt der Leistungsvergleich der Enzym-Substrat-Affinitätsregressionsaufgabe, der Enzym-Substrat-Paarklassifizierungsaufgabe und der Arzneimittel-Ziel-AffinitätsregressionsaufgabeIn den meisten Metriken übertrifft ESM-AA andere Modelle und erzielt hochmoderne Ergebnisse.Darüber hinaus übertrafen Feinabstimmungsstrategien (wie ProSmith und XGBoost), die auf ESM-AA basieren, durchweg die Versionen, die zwei unabhängige molekulare vortrainierte Modelle mit dem vortrainierten Proteinmodell kombinieren (wie in den letzten vier Zeilen der Tabellen 1 und 2 gezeigt).
Es ist erwähnenswert, dassESM-AA kann sogar Methoden schlagen, die vortrainierte Modelle mit größeren Parametergrößen verwenden.(Zum Beispiel der Vergleich zwischen der 5., 7. und letzten Zeile in Tabelle 2).
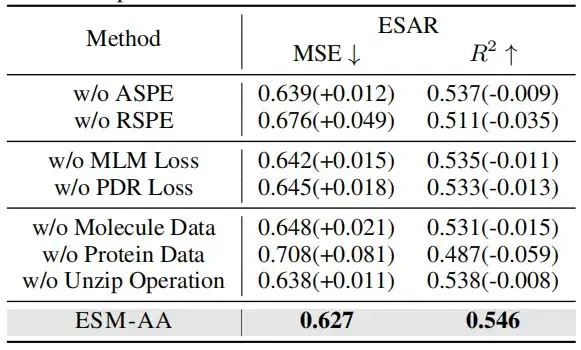
Um die Wirksamkeit der mehrskaligen Positionskodierung zu überprüfen, wurden in dieser Studie Ablationsexperimente in zwei Fällen durchgeführt: einer ohne Verwendung der atomar skaligen Positionskodierung (ASPE); die andere ist ohne Verwendung der genetischen Skalenpositionskodierung (RSPE).
Beim Entfernen von Molekülen oder Proteindaten sank die Modellleistung erheblich. Interessanterweise ist die Leistungsverschlechterung durch das Entfernen von Proteindaten deutlicher als die durch das Entfernen von molekularen Daten. Dies deutet darauf hin, dass das Modell, wenn es nicht anhand von Proteindaten trainiert wird, schnell proteinbezogenes Wissen verliert, was zu einem erheblichen Rückgang der Gesamtleistung führt. Jedoch,Auch ohne molekulare Daten kann das Modell durch Dekomprimierungsvorgänge Informationen auf atomarer Ebene erhalten.
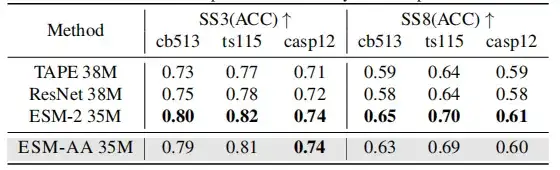
Da ESM-AA auf der Grundlage vorhandener PLMs entwickelt wurde, soll mit dieser Studie ermittelt werden, ob es noch immer ein umfassendes Verständnis von Proteinen aufweist. Dadurch soll die Fähigkeit vortrainierter Proteinmodelle zum Verständnis der Proteinstruktur durch die Verwendung von Aufgaben zur Vorhersage der Sekundärstruktur und zur Vorhersage unbeaufsichtigter Kontakte getestet werden.
Die Ergebnisse zeigen, dass ESM-AA zwar in dieser Art von Studie möglicherweise keine optimale Leistung erzielt,Seine Leistung bei der Vorhersage von Sekundärstrukturen und Kontakten ist jedoch ähnlich der von ESM-2.
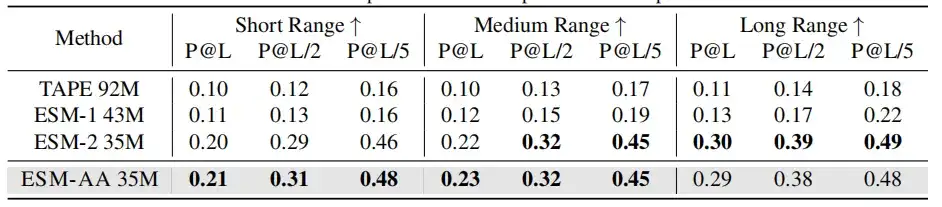
Beim molekularen BenchmarkingDie Leistung von ESM-AA ist bei den meisten Aufgaben mit der von Uni-Mol vergleichbar.Es übertrifft in vielen Fällen mehrere molekülspezifische Modelle und zeigt damit sein Potenzial als leistungsstarker Ansatz für molekulare Aufgaben.
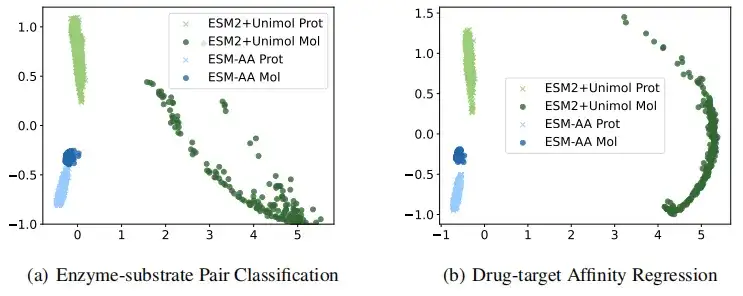
Um anschaulicher zu veranschaulichen, dass ESM-AA qualitativ hochwertigere Darstellungen von Proteinen und kleinen Molekülen liefert, wurden in dieser Studie die von ESM-AA und ESM-2+Uni-Mol extrahierten Darstellungen bei der Klassifizierung von Enzym-Substrat-Paaren und der Regression der Arzneimittelzielaffinität visuell verglichen. Die Ergebnisse zeigen, dassDas ESM-AA-Modell ist in der Lage, eine zusammenhängendere semantische Darstellung sowohl von Protein- als auch von Moleküldaten zu erstellen, wodurch ESM-AA die beiden separaten vortrainierten Modelle übertrifft.
Proteinsprachmodell, der nächste Schritt des großen Sprachmodells
Seit den 1970er Jahren sind immer mehr Wissenschaftler davon überzeugt, dass „das 21. Jahrhundert das Jahrhundert der Biologie ist“. Im vergangenen Juli schrieb Forbes einen langen Artikel, in dem es die Vorstellung hatte, dass der LLM Menschen an die Spitze einer neuen Runde von Veränderungen im Bereich der Biologie stellt. Die Biologie erweist sich als ein System, das entschlüsselt, programmiert und in gewisser Weise sogar digitalisiert werden kann.LLM bietet mit seiner erstaunlichen Fähigkeit, natürliche Sprachen zu steuern, das Potenzial zur Entschlüsselung biologischer Sprachen.Dies macht das Proteinsprachenmodell auch zu einem der beliebtesten Bereiche dieser Ära.
Proteinsprachmodelle stellen hochmoderne Anwendungen der KI-Technologie in der Biologie dar. Durch das Erlernen der Muster und Strukturen von Proteinsequenzen können die Funktion und Morphologie von Proteinen vorhergesagt werden, was für die Entwicklung neuer Medikamente, die Behandlung von Krankheiten und die biologische Grundlagenforschung von großer Bedeutung ist.
Zuvor haben Proteinsprachenmodelle wie ESM-2 und ESMFold eine mit AlphaFold vergleichbare Genauigkeit gezeigt, mit schnelleren Verarbeitungsgeschwindigkeiten und genaueren Vorhersagefähigkeiten für „verwaiste Proteine“. Dies beschleunigt nicht nur die Vorhersage der Proteinstruktur, sondern bietet auch neue Werkzeuge für das Protein-Engineering, sodass Forscher völlig neue Proteinsequenzen mit spezifischen Funktionen entwerfen können.
Darüber hinaus profitiert die Entwicklung von Proteinsprachenmodellen von den sogenannten „Skalierungsgesetzen“.Das heißt, die Leistung des Modells verbessert sich erheblich, wenn der Modellmaßstab, die Datensatzgröße und der Rechenaufwand zunehmen.Dies bedeutet, dass die Fähigkeiten des Proteinsprachenmodells mit der Erhöhung der Modellparameter und der Ansammlung von Trainingsdaten einen qualitativen Sprung machen werden.
In den letzten zwei Jahren haben Proteinsprachenmodelle auch in der Geschäftswelt eine Phase rasanter Entwicklung durchlaufen. Im Juli 2023 schlugen Baidu Biosciences und die Tsinghua-Universität gemeinsam ein Modell namens xTrimo Protein General Language Model (xTrimoPGLM) mit einer Parametergröße von bis zu 100 Milliarden (100B) vor, das bei mehreren Aufgaben zum Verständnis von Proteinen (13 von 15 Aufgaben) andere fortgeschrittene Basismodelle deutlich übertraf.Bei der Generierungsaufgabe ist xTrimoPGLM in der Lage, neue Proteinsequenzen zu generieren, die natürlichen Proteinstrukturen ähneln.
Im Juni 2024 gab das KI-Protein-Unternehmen Tushen Zhihe bekannt, dassDas erste natürlichsprachliche Proteinmodell in China, TourSynbio™, wurde allen Forschern und Entwicklern als Open Source zur Verfügung gestellt.Dieses Modell ermöglicht ein Verständnis der Proteinliteratur auf dialogorientierte Weise und umfasst Funktionen wie Proteineigenschaften, Funktionsvorhersage und Proteindesign. Im Hinblick auf die Bewertungsindikatoren beim Vergleich von Proteinbewertungsdatensätzen übertrifft es GPT4 und ist das erste in der Branche.
Darüber hinaus könnte der Durchbruch in der technologischen Forschung, den ESM-AA darstellt, auch bedeuten, dass die Technologieentwicklung kurz davor steht, den „Wright Brothers-Moment“ zu überwinden und einen großen Sprung nach vorne einzuleiten. Gleichzeitig wird die Anwendung von Proteinsprachenmodellen nicht nur auf die Bereiche Medizin und Biopharmazie beschränkt bleiben, sondern kann auch auf viele andere Bereiche wie Landwirtschaft, Industrie, Materialwissenschaften und Umweltsanierung ausgeweitet werden. Dies wird die technologische Innovation in diesen Bereichen fördern und beispiellose Veränderungen für die Menschheit mit sich bringen.








