Command Palette
Search for a command to run...
Das 100-Millionen-Parameter-Zellenmodell Ist Da! Team Der Tsinghua-Universität Veröffentlicht scFoundation Im Fachjournal „Nature“: Simultane Modellierung Von 20.000 Genen
In den letzten Jahren haben vortrainierte Modelle im großen Maßstab eine neue Welle der künstlichen Intelligenz angeführt. Das „große Modell“ extrahiert tiefgreifende Regeln aus umfangreichen Daten aus mehreren Quellen und kann dann als „Basismodell“ für eine Vielzahl von Aufgaben in unterschiedlichen Bereichen dienen. Große Sprachmodelle haben es beispielsweise geschafft, Sprache durch Lernen aus großen Mengen von Textdaten zu verstehen und zu erkennen und haben damit das Paradigma im Bereich der natürlichen Sprachverarbeitung revolutioniert.
Auch im Bereich der Biowissenschaften haben Organismen ihre eigene „zugrundeliegende Sprache“ – Zellen sind die grundlegenden strukturellen und funktionellen Einheiten des menschlichen Körpers.Werden DNA-, RNA-, Protein- und Genexpressionswerte zu „Wörtern“ verglichen, werden diese zu dem Satz „Zelle“ zusammengefügt.Wenn es uns also gelingt, ein Zellmodell mit künstlicher Intelligenz auf Grundlage der Zell-„Sprache“ zu entwickeln, wird dies hoffentlich ein neues Forschungsparadigma und revolutionäre Forschungsinstrumente für die Biowissenschaften und die Medizin schaffen.
Jedoch,Derzeit gibt es drei Hauptherausforderungen beim Training großer Einzelzelldaten:
* Vorabtrainingsdaten zur Genexpression müssen Zelllandschaften unterschiedlicher Zustände und Typen abdecken. Derzeit sind die meisten Daten zur Einzelzell-RNA-Sequenzierung (scRNA-seq) lose organisiert und es fehlt noch immer eine umfassende und vollständige Datenbank.
* Während des Trainings haben herkömmliche Transformer Schwierigkeiten, „Sätze“ zu verarbeiten, die aus fast 20.000 proteinkodierenden Genen bestehen;
* scRNA-seq-Daten aus verschiedenen Technologien und Laboren unterscheiden sich in der Sequenzierungstiefe, was das Modell daran hindert, einheitliche und aussagekräftige Zell- und Gendarstellungen zu erlernen.
Um diese Herausforderungen zu bewältigen,An der Forschung arbeiteten Professor Zhang Xuegong, Direktor des Life Basic Model Laboratory der Abteilung für Automatisierung der Tsinghua-Universität, Professor Ma Jianzhu von der Abteilung für Elektronik/AIR und Dr. Song Le von der Abteilung Biotechnologie zusammen.Im Juni 2024 wurde in Nature Methods eine Forschungsarbeit mit dem Titel „Large-scale foundation model on single-cell transcriptomics“ veröffentlicht.
Das Papier stellt ein Großzellmodell namens scFoundation vor, das etwa 20.000 Gene gleichzeitig verarbeiten kann.Als Basismodell weist es herausragende Leistungsverbesserungen bei einer Vielzahl von nachgelagerten biomedizinischen Aufgaben auf, beispielsweise bei der Verbesserung der Zellsequenzierungstiefe, der Vorhersage der Zellreaktion auf Arzneimittel und der Vorhersage von Zellstörungen, und bietet damit ein neues Paradigma für künstliche Intelligenz in der Einzelzellforschung.
Forschungshighlights:
Das scFoundation-Zellmodell wird anhand von Genexpressionsdaten von 50 Millionen Zellen trainiert, verfügt über 100 Millionen Parameter und kann ungefähr 20.000 Gene gleichzeitig verarbeiten.* Das Modell verwendet ein asymmetrisches Design, um den Rechen- und Speicheraufwand zu reduzieren.* Das Modell liefert neue Forschungsideen für die Inferenz von Gennetzwerken und die Identifizierung von Transkriptionsfaktoren.

Papieradresse:
https://www.nature.com/articles/s41592-024-02305-7
Das Open-Source-Projekt „awesome-ai4s“ vereint mehr als 100 AI4S-Papierinterpretationen und stellt außerdem umfangreiche Datensätze und Tools bereit:
https://github.com/hyperai/awesome-ai4s
Datensätze: Erstellen eines umfassenden Einzelzelldatensatzes
Die Forscher erstellten einen umfassenden Einzelzell-Datensatz, indem sie alle öffentlich verfügbaren Einzelzell-Ressourcendaten sammelten.Dazu gehören Gene Expression Omnibus (GEO), Single Cell Portal, HCA, Human Genome Project (hECA), Deeply Integrated Human Single-Cell Omics Data (DISCO), Datenbank des Europäischen Labors für Molekularbiologie – Europäisches Bioinformatikinstitut (EMBL-EBI) usw.
* GEO-Download-Adresse:https://www.ncbi.nlm.nih.gov/geo/
* Download-Link für das Single Cell Portal:https://singlecell.broadinstitute.org/single_cell
* HCA-Downloadadresse:https://data.humancellatlas.org/
* EMBL-EBI-Downloadadresse:https://www.ebi.ac.uk/
Die Forscher glichen alle Daten mit einer Genliste von 19.264 proteinkodierenden und häufigen mitochondrialen Genen ab, die vom HUGO Gene Nomenclature Committee identifiziert wurden. Nach der DatenqualitätskontrolleFür das Vortraining wurden mehr als 50 Millionen menschliche scRNA-Sequenzdaten beschafft.
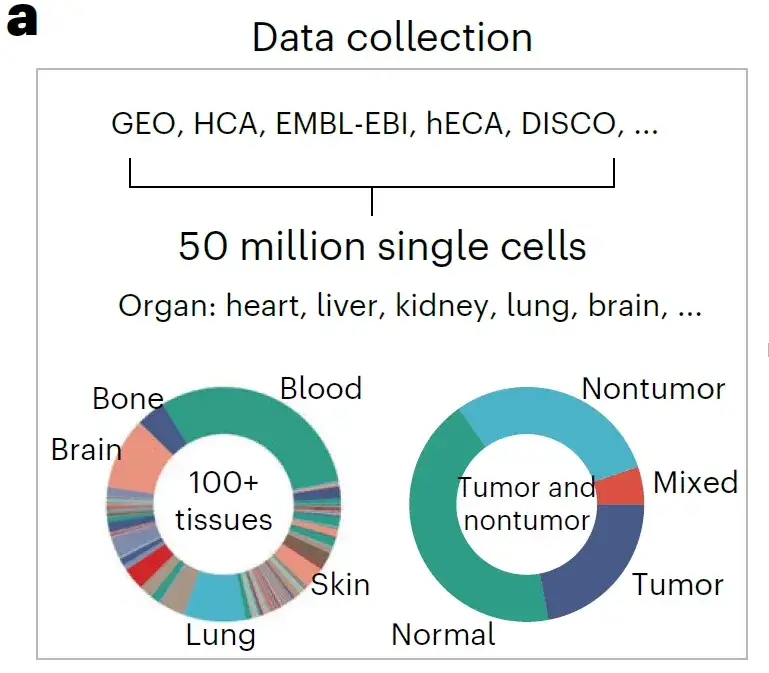
Zahlreiche Datenquellen erstellen vor dem Training Datensätze, die reich an biologischen Mustern sind. Anatomisch gesehen umfasst es mehr als 100 Gewebetypen und deckt ein breites Spektrum an Krankheiten, Tumoren und Normalzuständen ab. Wie in der Abbildung oben gezeigt, umfasst es fast alle bekannten menschlichen Zelltypen und -zustände.
Modellarchitektur: Erstellen eines scFoundation-Modells mit 100 Millionen Parametern
Das von den Forschern entwickelte scFoundation-Modell verfügt über etwa 100 Millionen Parameter und seine Parameterskala, Genabdeckung und Datenskala gehören „zu den besten“ im Einzelzellbereich.
Modelldesign
Die Forscher entwickelten das xTrimoGene-Modell als Backbone-Modell von scFoundation, einem skalierbaren, auf Transformer basierenden Modell, das ein Einbettungsmodul und eine asymmetrische Encoder-Decoder-Struktur umfasst.
Unter diesen konvertiert das Vektormodul kontinuierliche Skalarwerte der Genexpression in lernbare hochdimensionale Vektoren und stellt sicher, dass die ursprünglichen Expressionswerte vollständig erhalten bleiben. Der Encoder verwendet als Eingabe nicht-null und nicht maskierte Expressionsgene, verwendet einen Vanilla-Transformerblock und verfügt über eine große Anzahl von Parametern. Der Decoder verwendet alle Gene als Eingabe, verwendet einen Performer-Block und hat eine relativ geringe Anzahl von Parametern.
Dieses asymmetrische Design reduziert den Rechen- und Speicheraufwand im Vergleich zu anderen Architekturen.Die Daten zeigen, dass das Modul nur 3,4% des traditionellen Sprachmodells Transformer erfordert und dabei die gleiche Parameterskala beibehält.
Vorschulungsaufgaben
Die Forscher entwickelten eine Vortrainingsaufgabe namens RDA-Modellierung (Read-Depth-Aware).Dies ist eine Erweiterung des maskierten Sprachmodells, die die hohe Varianz der Sequenzierungstiefe in großen Datenmengen berücksichtigt.
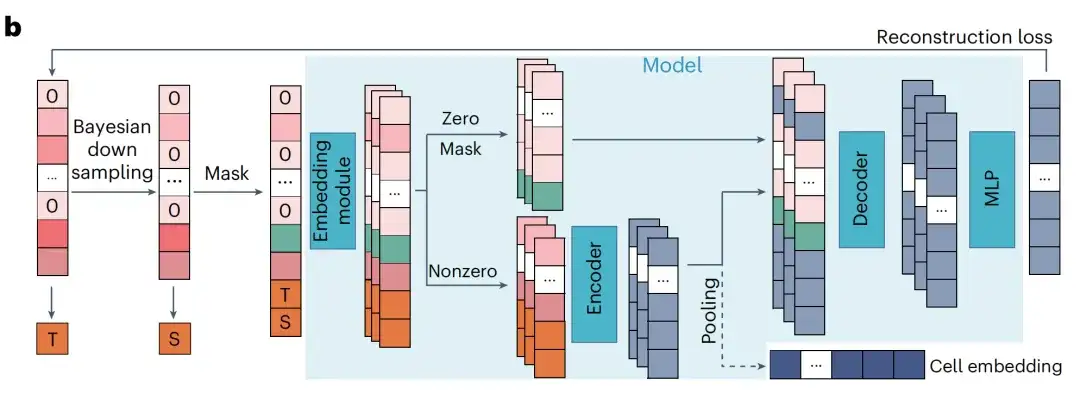
Bei der RDA-Modellierung sagt das Modell die Expression maskierter Gene basierend auf den Kontextgenen der Zelle voraus. Die Forscher betrachteten die Gesamtzählung als Sequenzierungstiefe einer Zelle und definierten zwei Metriken für die Gesamtzählung: T (Ziel) und S (Quelle), die jeweils die Gesamtzählungen der Originalprobe und der Eingabeprobe darstellen. Die Forscher maskierten in den Eingabeproben zufällig Null-exprimierte und Nicht-Null-exprimierte Gene und zeichneten ihre Indizes auf.
Das Modell verwendet dann die maskierte Eingabestichprobe und die beiden Metriken, um den Ausdruckswert der ursprünglichen Stichprobe am maskierten Index vorherzusagen. Dadurch kann das vortrainierte Modell nicht nur genetische Beziehungen innerhalb von Zellen erfassen, sondern auch Zellen unterschiedlicher Sequenzierungstiefe koordinieren. Während der Inferenz geben die Forscher die Rohgenexpression von Zellen in das vortrainierte Modell ein und setzen T höher als ihre Gesamtzählungen S, um Genexpressionswerte mit erhöhter Sequenzierungstiefe zu erzeugen.
Einfach ausgedrückt kann RDA die Sequenzierungstiefe heruntersampeln, sodass das Modell zusätzlich zur Erledigung der herkömmlichen Maskenwiederherstellungsaufgabe in der Vortrainingsphase auch die Genexpressionsdaten hochwertiger Zellen aus minderwertigen Zellen wiederherstellen kann.
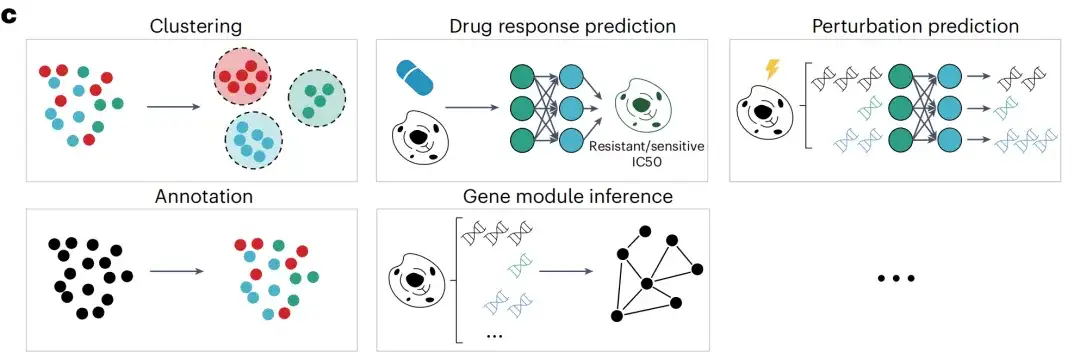
Nach dem Vortraining wendeten die Forscher das scFoundation-Modell auf mehrere nachgelagerte Aufgaben an. Die Ausgaben der scFoundation-Encoder werden in Vektoren auf Zellebene zusammengefasst, um sie in Aufgaben auf Zellebene zu verwenden, darunter Clustering (innerhalb und über Datensätze hinweg), Vorhersage der Arzneimittelreaktion auf Batch- und Einzelzellebene sowie Zelltypannotation. Die Ausgabe des scFoundation-Decoders ist ein Kontextvektor auf Genebene, der für Aufgaben auf Genebene wie Störungsvorhersage und Genmodul-Inferenz verwendet wird.
Forschungsergebnisse: scFoundation-Modelle weisen eine hervorragende Leistung auf
In tatsächlichen Anwendungen unterstützt das scFoundation-Modell zwei Modi: „Out-of-the-Box“ und „Feintuning“.Im „Out-of-the-Box“-Modus kann das Modell dank seiner einzigartigen Vortrainingsaufgaben direkt zur Verbesserung der Qualität von Zelldaten verwendet werden und ohne weitere Anpassung dieselben oder bessere Ergebnisse erzielen als bestehende Methoden. Darüber hinaus können Benutzer mit scFoundation vortrainierte Darstellungen von Zellen extrahieren, die zur Identifizierung zelltypspezifischer Genmodule und Transkriptionsfaktoren verwendet werden können und in nachgelagerten Aufgaben umfassend eingesetzt werden können.
Skalierbares, feinabstimmungsfreies Sequenzierungs-Deep-Enhancement-Modell
Die Forscher trainierten drei Modelle mit 3M, 10M und 100M Parametern und zeichneten ihre Verluste im Validierungsdatensatz auf.
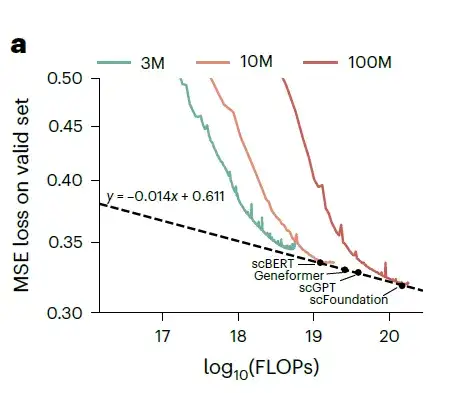
Trainingsverlust bei unterschiedlichen Parametergrößen und FLOPs. Die grüne Kurve stellt das 3M-Modell dar, die orange Kurve das 10M-Modell und die rote Kurve das 100M-Modell.
Mit zunehmenden Modellparametern und Gleitkommaoperationen (FLOPs) nimmt der Verlust im Validierungsdatensatz potenzmäßig ab. Anschließend schätzten die Forscher die Leistung von xTrimoGene-Architekturmodellen verschiedener Größen und verglichen sie mit scVI. Wie in der Abbildung oben gezeigt,Das scFoundation-Modell mit 100 Millionen Parametern schnitt von allen Modellen am besten ab.Die Forscher bewerteten die drei Modelle außerdem anhand der Zelltyp-Annotationsaufgabe und stellten fest, dass sich die Leistung mit zunehmender Modellgröße verbesserte.
Die Forscher bewerteten diese Fähigkeit anhand eines unabhängigen Testdatensatzes von 10.000 Zellen, die zufällig aus dem Validierungsdatensatz ausgewählt wurden, indem sie die Gesamtzählungen auf 1%, 5%, 10% und 20% der Originaldaten heruntersampelten und so vier Datensätze mit unterschiedlichen Variationen der Gesamtzählung erstellten. Für jeden Datensatz wurden der mittlere absolute Fehler (MAE), der mittlere relative Fehler (MRE) und der Pearson-Korrelationskoeffizient (PCC) zwischen den vorhergesagten Werten und der tatsächlichen, von Null verschiedenen Genexpression mithilfe von nicht abgestimmtem scFoundation gemessen.
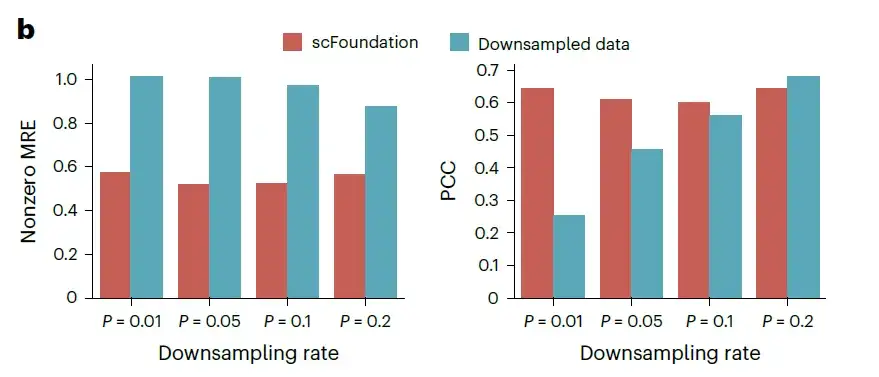
Bewertung der Leistung der Lesetiefenverbesserung bei unbekannten Datensätzen MRE und PCC wurden verwendet, um die Leistung der wiederhergestellten Genexpression zu bewerten, wobei niedrigere MRE und höhere PCC eine bessere Leistung anzeigen.
Wie in der obigen Abbildung gezeigt, werden MAE und MRE von scFoundation selbst dann deutlich um die Hälfte reduziert, wenn die Downsampling-Rate niedriger als 10% ist.Diese Ergebnisse zeigen die Fähigkeit von scFoundation, die Genexpression bei extrem niedrigen Gesamtzahlen zu steigern.
Nachgelagerte Aufgaben – Aufgaben zur Vorhersage der Wirkung von Krebsmedikamenten
Ziel der Krebsmedikamentenreaktionen (CDRs) ist die Untersuchung der Reaktionen von Tumorzellen auf medikamentöse Eingriffe. Die computergestützte Vorhersage von CDRs ist für die Entwicklung von Krebsmedikamenten und das Verständnis der Krebsbiologie von entscheidender Bedeutung. In dieser Studie kombinierten die Forscher scFoundation mit der CDR-Vorhersagemethode DeepCDR, um den IC50-Wert der halbmaximalen Hemmkonzentration von Arzneimitteln in Daten mehrerer Zelllinien vorherzusagen und zu überprüfen, ob scFoundation nützliche Einbettungsinformationen für allgemeine Genexpressionsdaten basierend auf Einzelzelltraining liefern kann.
Die Forscher bewerteten die Leistung der auf scFoundation basierenden Ergebnisse im Vergleich zu den auf Genexpression basierenden Ergebnissen für mehrere Medikamente und Krebszelllinien.Die Ergebnisse zeigten, dass die meisten Medikamente und alle Krebsarten durch die scFoundation-Einbettung höhere Pearson-Korrelationskoeffizienten (PCC) erreichten.Wie in der folgenden Abbildung dargestellt:
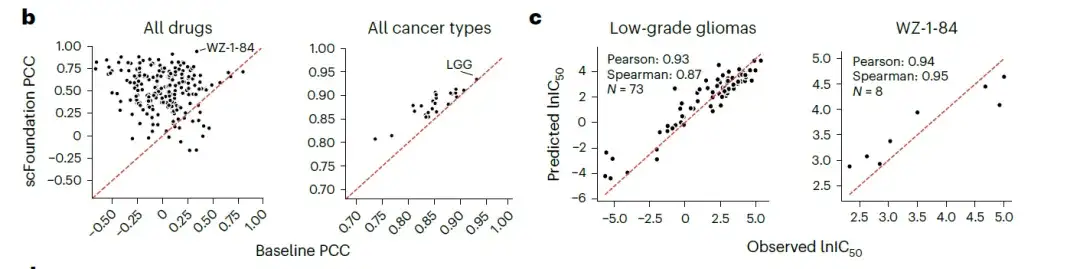
- Hinweis: Der Pearson-Korrelationskoeffizient ist eine Statistik, die die Stärke der linearen Beziehung zwischen Variablen misst, und sein Wertebereich liegt zwischen -1 und 1. Wenn der Korrelationskoeffizient nahe bei 1 liegt, weist dies darauf hin, dass zwischen den beiden Variablen eine vollständig positive lineare Beziehung besteht; wenn es nahe bei -1 liegt, zeigt dies an, dass eine vollständig negative lineare Beziehung vorliegt; Wenn der Wert nahe 0 liegt, bedeutet dies, dass zwischen den beiden Variablen keine lineare Beziehung besteht.
Dies zeigt, dassObwohl scFoundation anhand von Einzelzell-Transkriptomdaten vortrainiert ist, können die erlernten Genbeziehungen auf globale Expressionsdaten übertragen werden.Generiert einen komprimierten Vektor, der genauere IC50-Vorhersagen ermöglicht. Daher verfügt die scFoundation über das Potenzial, unser Verständnis der Arzneimittelreaktionen in der Krebsbiologie zu erweitern und die Entwicklung wirksamerer Krebsbehandlungen zu unterstützen.
Nachgelagerte Aufgabe – Klassifizierungsaufgabe für die Reaktion einzelner Zellen auf Arzneimittel
Durch die Bestimmung der Arzneimittelempfindlichkeit auf Einzelzellebene können bestimmte Zellsubtypen identifiziert werden, die unterschiedliche Arzneimittelresistenzprofile aufweisen. Dies liefert wertvolle Einblicke in die zugrunde liegenden Mechanismen und ermöglicht neue Therapieansätze. Daher wandten die Forscher scFoundation auf die kritische Aufgabe der Klassifizierung der Arzneimittelreaktion einzelner Zellen an, basierend auf einem nachgelagerten Modell namens SCAD.
Die Forscher konzentrierten sich auf vier Medikamente (Sorafenib, NVP-TAE684, PLX4720 und Etoposid), die in den ursprünglichen Studien niedrigere AUC-Werte (Area Under Curve) zeigten. Das auf scFoundation basierende Modell wurde mit dem Basis-SCAD-Modell verglichen, das alle Genexpressionswerte als Eingabe verwendete. Die Ergebnisse zeigten, dass das auf scFoundation basierende Modell bei allen Medikamenten höhere Werte bei den AUC-Werten erreichte, insbesondere bei NVP-TAE684 und Sorafenib, wobei die AUC-Werte um mehr als 0,2 anstiegen, wie in der folgenden Abbildung dargestellt.
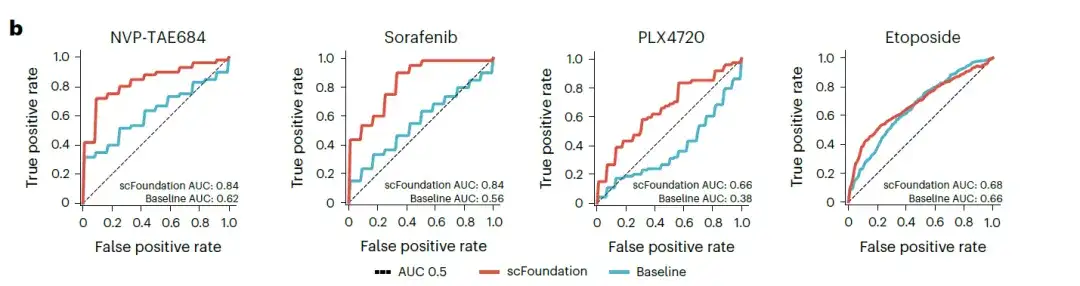
Hinweis: AUC kann verwendet werden, um die Leistung des Modells zu messen. Der Wertebereich von AUC liegt zwischen 0 und 1. Je größer der Wert, desto besser ist die Klassifizierungsleistung des Modells.
Diese Ergebnisse bestätigen das Potenzial der Verwendung der scFoundation-Einbettung zur Erfassung von Biomarkersignalen zur Arzneimittelempfindlichkeit.
Nachgelagerte Aufgabe – Aufgabe zur Vorhersage von Zellstörungen
Das Verständnis der zellulären Reaktionen auf Störungen ist für biomedizinische Anwendungen und die Arzneimittelentwicklung von entscheidender Bedeutung, da es bei der Identifizierung von Gen-Gen-Interaktionen und potenziellen Angriffspunkten für Arzneimittel in verschiedenen Zelltypen hilft. Die Forscher kombinierten scFoundation mit einem fortschrittlichen Modell, GEARS, um Störungsreaktionen mit Einzelzellauflösung vorherzusagen, und berechneten als Bewertungskriterium den durchschnittlichen mittleren quadratischen Fehler (MSE) der 20 am stärksten unterschiedlich exprimierten (DE) Gene mit signifikant unterschiedlichen Genexpressionsprofilen vor und nach der Behandlung.
Die Ergebnisse zeigen, dassIm Vergleich zum ursprünglichen GEARS-Basismodell erreichte das auf scFoundation basierende Modell niedrigere MSE-Werte.Die folgende Abbildung zeigt die Expressionsänderungen der 20 wichtigsten Gene bei der doppelten Genstörung ETS2 + CEBPE:
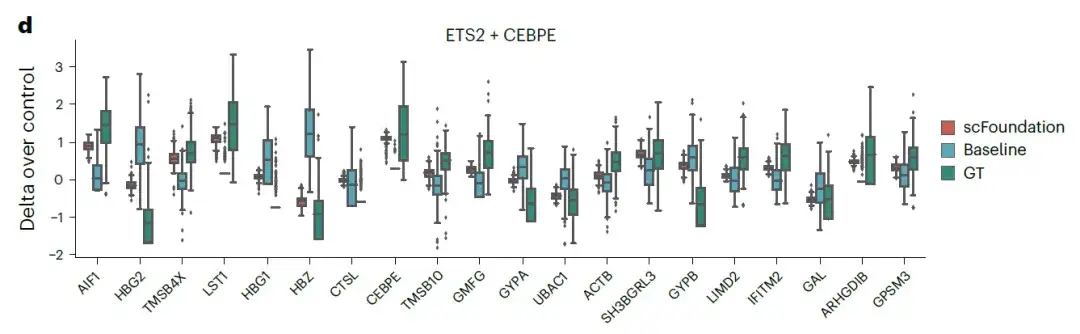
Diese Ergebnisse legen nahe, dass durch die Extraktion von Gendarstellungen aus einzelnen Zellen zur Konstruktion spezifischer Gen-KoexpressionsnetzwerkescFoundation hat die Zell- und Geneigenschaften unter verschiedenen Bedingungen erfolgreich erfasst und die Genauigkeit der Vorhersagen einzelner/doppelter Störungen deutlich verbessert.
Zusammenfassend lässt sich sagen, dass das scFoundation-Modell neue Ideen und Methoden für die Erstellung der Modellarchitektur, des Trainingsrahmens und des nachgelagerten Demonstrationsanwendungssystems für Vortrainingsmodelle großer Zellen bietet, grundlegende Funktionen für das Erlernen biomedizinischer Aufgaben bereitstellt und die Grenzen grundlegender Modelle im Einzelzellbereich erweitert.
Erforschung großer Life-Science-Modelle mit besserer Leistung
Demis Hassabis, CEO und Gründer von DeepMind, einem weltweit führenden Unternehmen für künstliche Intelligenz, sagte einmal:„Grundsätzlich kann die Biologie als ein sehr komplexes und dynamisches System der Informationsverarbeitung betrachtet werden. So wie sich die Mathematik als die richtige Beschreibungssprache für die Physik erwiesen hat, könnte die Biologie ein ideales Anwendungsgebiet für künstliche Intelligenz sein.“
Allerdings erfordern herkömmliche KI-Methoden große Mengen gekennzeichneter Daten, um genaue Vorhersagen zu treffen. Doch in den Biowissenschaften sind qualitativ hochwertige, gekennzeichnete Daten oft Mangelware. Der Wunsch, genauere Downstream-Task-Modelle auf der Grundlage einer geringeren Datenmenge zu erstellen, bedeutet, dass das zugrunde liegende Basismodell über eine bessere Darstellung oder allgemeine Funktionen verfügen muss. Aus diesem Grund arbeiten immer mehr Forscher an der Entwicklung besserer vertikaler Makromodelle im biologischen Bereich.
Mai 2023Ein Forschungsteam der Universität Toronto hat das erste groß angelegte Sprachmodell auf Basis der Einzelzellbiologie veröffentlicht: scGPT.Das Modell wurde auf über 10 Millionen Zellen vortrainiert und ermöglicht Transferlernen für verschiedene nachgelagerte Aufgaben. Im Juli desselben Jahres versuchte das Team außerdem, scGPT zu aktualisieren, indem es ein Vortraining auf mehr als 33 Millionen Zellen durchführte. Die Ergebnisse zeigten, dass scGPT wichtige biologische Erkenntnisse über Gene und Zellen effektiv extrahieren und bei verschiedenen nachgelagerten Aufgaben eine verbesserte Leistung erzielen kann, darunter Multi-Batch-Integration, Multi-Omics-Integration, Zelltyp-Annotation, Vorhersage genetischer Störungen und Gennetzwerk-Inferenz.
Die Studie mit dem Titel „scGPT: Auf dem Weg zum Aufbau eines Grundlagenmodells für Einzelzell-Multiomik mithilfe generativer KI“ wurde in Nature Methods veröffentlicht.
* Link zum Artikel:https://www.nature.com/articles/s41592-024-02201-0
September 2023Dem Xcompass-Konsortium, einem multidisziplinären Forschungsteam der Chinesischen Akademie der Wissenschaften, ist es gelungen, das weltweit erste artenübergreifende Lebensgrundlagenmodell zu entwickeln: GeneCompass.Das Modell integriert Transkriptomdaten von mehr als 126 Millionen Einzelzellen von Menschen und Mäusen, vereint vier Arten von Vorwissen, darunter Promotorsequenzen und Gen-Koexpressionsbeziehungen, und verfügt über 130 Millionen grundlegende Modellparameter, wodurch ein umfassendes Lernen und Verständnis der Gesetze der Genexpressionskontrolle erreicht wird und gleichzeitig die Vorhersage von Zellzustandsänderungen und die genaue Analyse verschiedener Lebensprozesse unterstützt werden.

Die Studie wurde auf bioRxiv unter dem Titel „GeneCompass: Deciphering Universal Gene Regulatory Mechanisms with Knowledge-Informed Cross-Species Foundation Model“ veröffentlicht.
- Link zum Artikel:https://www.biorxiv.org/content/10.1101/2023.09.26.559542v1
Im Oktober 2023 gab der globale Pharmariese Sanofi eine groß angelegte strategische Partnerschaft mit BioMap BioScience bekannt. Die beiden Parteien werden gemeinsam hochmoderne Modelle für die Entdeckung biotherapeutischer Arzneimittel entwickeln, die auf dem Life Science AI Foundation Model von BioMap basieren.
Mit Blick auf die Zukunft wird die Anwendung des komplexen Verständnisses und der innovativen Generierungsfähigkeiten großer Sprachmodelle, die die menschliche Vorstellungskraft weit übersteigen, auf die komplexere „natürliche Sprache“ des Lebens hoffentlich das Forschungsparadigma der Biowissenschaften wirklich verändern.
Quellen:
1.https://www.jiqizhixin.com/articles/2023-9-29
2.https://www.tsinghua.edu.cn/info/1175/112118.htm
3.https://hope.huanqiu.com/article/4FYZxnpu88J
4.https://www.jiqizhixin.com/articles/2023-7-5-26'








