Command Palette
Search for a command to run...
Koreanische Version Von AlphaFold? Deep-Learning-Modell AlphaPPIMd: Zur Ensemble-Erkundung Von Protein-Protein-Komplex-Konformationen

Protein spielt auf der Bühne des Lebens eine unverzichtbare Rolle. Sie sind die aktivsten Moleküle in lebenden Organismen und sind am Zellaufbau, an der Reparatur, an der Energieumwandlung, an der Signalübertragung und an zahllosen wichtigen biologischen Funktionen beteiligt. Gleichzeitig ist die Struktur von Proteinen eng mit ihren Funktionen verbunden und ihre Funktionen werden durch komplexe Wechselwirkungen mit Proteinen, Peptiden, Nukleotiden und verschiedenen kleinen Molekülen erreicht. Diese Protein-Protein-Interaktion (PPI) ist der Kern vieler biologischer Prozesse innerhalb von Zellen, von der Zellsignalisierung über Immunreaktionen bis hin zur Regulierung des Zellzyklus.
Unser derzeitiges Verständnis der dreidimensionalen Struktur von Proteinen und ihrer Interaktionseigenschaften ist jedoch noch unvollständig. Traditionelle experimentelle Techniken wie Röntgenkristallographie und Kryo-Elektronenmikroskopie,Obwohl es hochauflösende Informationen zur Proteinstruktur liefern kann, ist es zeitaufwändig und kostspielig.Darüber hinaus stehen sie vor Herausforderungen bei der Analyse dynamischer Prozesse und selten vorkommender Proteine. Dies schränkt das tiefgreifende Verständnis der Proteinfunktionen und Interaktionsmechanismen erheblich ein und beeinträchtigt wiederum die Entwicklung des Arzneimitteldesigns und der Proteintechnik.
Um dieses Problem zu lösen, kombinierten Dr. Jianmin Wang von der Yonsei-Universität und seine Mitarbeiter Deep Learning mit generativer KI.Verwendung von Transformer-basiertem generativem neuronalen Netzwerklernen zur Erforschung des Konformationsensembles von Protein-Protein-Komplexen,Die Schlüsselrückstände, die die Konformation und Dynamik von Protein-Protein-Komplexen beeinflussen, wurden aus mehreren molekulardynamischen (MD)-Trajektorien ermittelt und lieferten mechanistische Einblicke in die Protein-Protein-Bindung.

Papieradresse:
https://doi.org/10.1101/2024.02.24.581708
AlphaPPIMd-Modell: Basierend auf molekularer Dynamiksimulation, mit dem Selbstaufmerksamkeitsmechanismus als Kern
Das Forschungsteam verwendete den komplexen Barnase-Barstar-Trajektoriensatz als Datensatz.Zunächst wurde die Kristallstruktur des Barnase-Barstar-Komplexes aus der Protein Data Bank (PDB) heruntergeladen und die A- und D-Ketten durch Entfernen des Liganden und des kristallographischen Wassers als anfängliche Komplexstruktur extrahiert. Anschließend fügten die Forscher die fehlenden Wasserstoffatome mithilfe des tleap-Moduls in AmberTools hinzu und neutralisierten sie durch Zugabe von Na+- und Cl--Ionen, wobei sie diese innerhalb einer 12Å großen periodischen Begrenzungsbox von TIP3P-Wassermolekülen lösten. Schließlich wurden die Topologie- und Koordinatendateien des Systems mithilfe des Tleap-Moduls in AmberTools und dem AMBER ff14SB-Kraftfeld kompiliert.
Anschließend führte das Forschungsteam mithilfe eines Molekulardynamik-Simulationssystems 500 Schritte typischer NVT-Simulationen durch und verwendete dabei einen Langevin-Integrator, um die Energie zu minimieren. Anschließend wurden 10.000 Schritte einer NPT-Simulation bei 300 K durchgeführt, um den Gleichgewichtszustand weiter zu erreichen. Außerdem wurde der Partikelnetzwerk-Ewald-Algorithmus verwendet, um die elektrostatischen Wechselwirkungen über große Entfernungen zu berechnen. Der Grenzwert für direkte räumliche Wechselwirkungen wurde auf 1 nm festgelegt, der Simulationszeitschritt auf 2 fs und der SHAKE-Algorithmus wurde so eingestellt, dass die Länge aller Bindungen mit Wasserstoffatomen begrenzt wird. Anschließend wurden sechs unabhängige 100-ns-Moleküldynamiksimulationen durchgeführt. Alle Simulationen wurden mit OpenMM 7.7 durchgeführt.
Nach Abschluss der molekulardynamischen SimulationDas Forschungsteam erstellte das AlphaPPIMd-Modell auf Basis von Transformer und verwendete dabei ein tiefes generatives Modell, um Proteinkonformationszustände zu erfassen, die mit herkömmlicher Molekulardynamik nur schwer zu analysieren sind. Der Kern des AlphaPPImd-Frameworks ist der Self-Attention-Mechanismus, der die wichtigsten Aminosäurerestpaare erfassen kann, die die Konformation von Protein-Protein-Komplexen aus MD-Trajektorien beeinflussen.
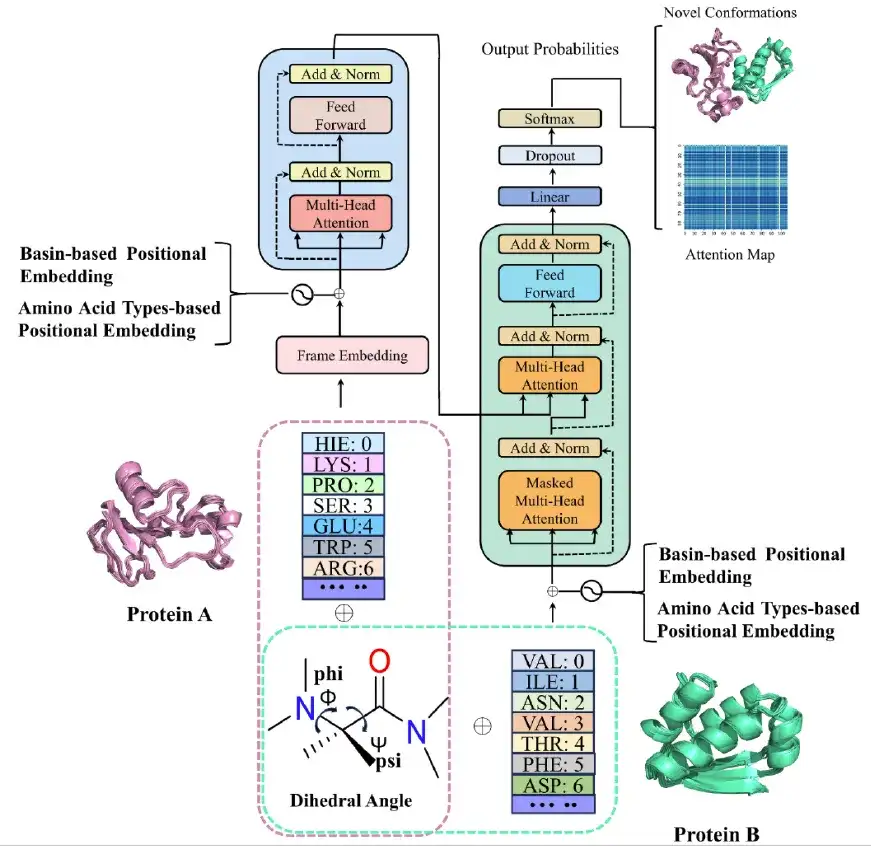
Erste,Das AlphaPPImd-Framework verarbeitet die MD-Trajektorie des Protein-Protein-Komplexes vor, um die Sequenzlänge, Sequenzzusammensetzung und den Aminosäureresttyp der beiden Ketten zu erhalten, und berechnet die Φ,Ψ-Winkel ausgewählter Reste in der Trajektorie, um verschiedene Konformationszustände darzustellen. (Wie in den rosa und grün gepunkteten Kästchen im obigen Bild gezeigt)
Zweitens,Die Forscher geben jedes Bild der MD-Trajektorie des Protein-Protein-Komplexes über das Einbettungsmodul in das Encodermodul von AlphaPPImd ein, das einen Mehrkopf-Selbstaufmerksamkeitsmechanismus, einen Aufmerksamkeitswert und ein Merkmalsoptimierungsmodul enthält. Der Decoder von AlphaPPImd wird verwendet, um die Beiträge von Resten verschiedener Typen und Positionen in einem Proteinkomplex zur Konformation zu lernen und zu erfassen.
endlich,Das Vorhersagemodul generiert iterativ den Grundzustand für das nächste Frame, und Modeller kann das Konformationsmodell des Protein-Protein-Komplexes basierend auf der erweiterten Grundzustands-Kodierungstrajektorie rekonstruieren.
Die Multi-Head-Self-Attention-Schicht im AlphaPPImd-Decodermodul lernt die Interaktionen zwischen bestimmten Restpaaren. Die Aufmerksamkeitsfunktion kann als Zuordnung zwischen der Abfrage (Q) und der Schlüssel-Wert-Ausgabe (KV) betrachtet werden. AlphaPPImd verwendet Proteinkomplex-Resteeinbettungen als Q, globale Proteinkomplex-Merkmale als K und V und berechnet Aufmerksamkeitsgewichte unter Verwendung von Q und K. Die Berechnungsformel lautet wie folgt:
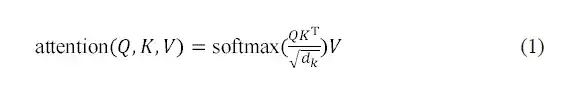
Gleichzeitig unterteilte die Studie sechs unabhängige 100-ns-MD-Trajektorien des Barnase-Barstar-Komplexes in 300 Primitive, die jeweils aus 1.000 Frames bestanden. Die MD-Trajektorien wurden vorverarbeitet, um nur Proteinatome beizubehalten. Jeder MD-Lauf liefert eine begrenzte Anzahl physikalischer Schnappschüsse von Protein-Protein-Komplexen. Jeder Frame in der Trajektorie wird als Grundzustand der Φ,Ψ-Kodierung dargestellt. daher,Der Torsionszustand eines Protein-Protein-Komplexes wird auf eine Textdarstellung reduziert,Die wesentlichen Nebenmerkmale der Dynamik bleiben erhalten.
Forschungsergebnis: Die durchschnittliche Trainingsgenauigkeit liegt bei bis zu 0,995 und kann auf weitere Proteinkomplexe ausgeweitet werden.
Der Barnase-Barstar-Komplex besteht aus zwei verschiedenen Ketten mit insgesamt 197 Resten (Barnase-Kette: 108 Reste, Barstar-Kette: 89 Reste). In der Studie wurde der KMeans-Algorithmus verwendet, um die Standorte in vier Cluster mit der Bezeichnung 0 (lila in der Abbildung unten), 1 (dunkelblau in der Abbildung unten), 2 (grün in der Abbildung unten) und 3 (gelb in der Abbildung unten) aufzuteilen. Anschließend wurde der Schwerpunkt jedes Clusters aufgezeichnet und gespeichert, um das Allatommodell des Barnase-Barstar-Komplexes aus dem im Grundzustand kodierten Torsionszustand zu rekonstruieren.
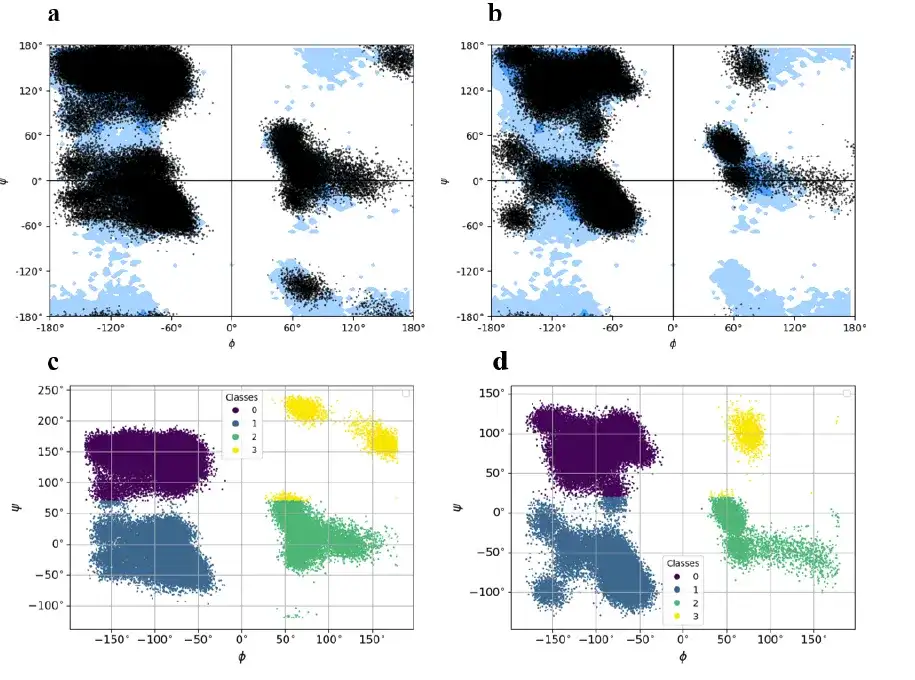
Diese Studie wandelt die Flugbahn jedes Frames in einen Zeichenvektor um, von denen jeder aus 4 Symbolen besteht, die 4 Clustern entsprechen. Schließlich wurde ein ähnlicher Darstellungsprozess für alle 300 Motive im MD-Trajektorien-Datensatz des Barnase-Barstar-Komplexes durchgeführt.
Zusammenfassend:Der Barnase-Barstar-Komplex ist ein Heterodimer mit deutlichen Unterschieden in den Grundzuständen der in den beiden Ketten kodierten Reste.Dies impliziert, dass sich der Barnase-Barstar-Komplex bei der Generierung neuer Grundzustands-Kodierungsrahmen und bei der Rekonstruktion von Konformationsmodellen einzelner Proteine erheblich unterscheidet.
Untersuchungen zeigen, dassDie durchschnittliche Trainingsgenauigkeit des AlphaPPImd-Modells beträgt 0,995 und die durchschnittliche Validierungsgenauigkeit 0,999.Obwohl AlphaPPImd schnell eine stabile Leistung erreichte, wurden in dieser Studie mehrere MD-Trajektorien als Datensätze verwendet, um das Transformer-Modell weiter zu verbessern und die vom Modell erlernte MD-Konformationsverteilung zu bereichern. Beispielsweise wurde in der Studie zufällig ein Frame aus der Trajektorie des Testsatzes als Eingabe ausgewählt und das trainierte AlphaPPImd-Framework verwendet, um 100 Grundzustands-Kodierungsframes zu generieren.
Die Ergebnisse zeigen, dassDas Modell ist in der Lage, Konformationen erfolgreich abzutasten und zu entfalten.Und die Diederbeschränkungen von Φ und Ψ können korrekt durchgesetzt werden.
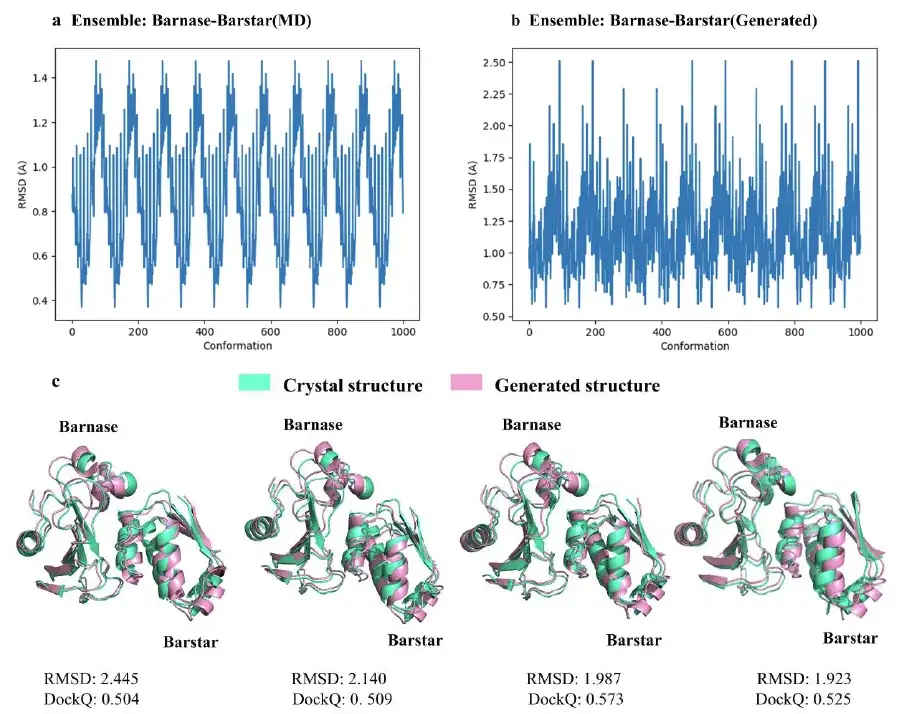
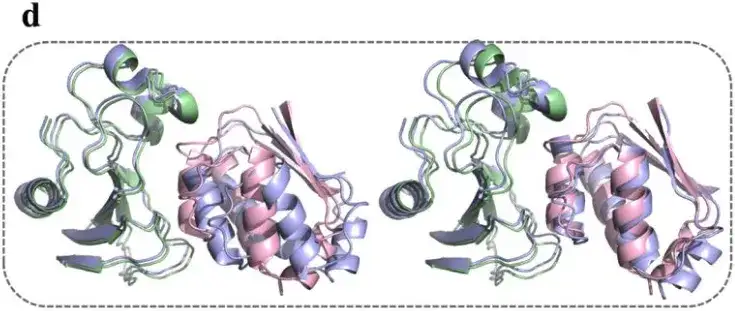
Die Studie wählte außerdem vier repräsentative Konformationen mit RMSD nahe 2Å aus den 1.000 Barnase-Barstar-Komplex-Konformationen aus, die vom AlphaPPImd-Modell generiert wurden. Die Ergebnisse der Studie zeigten, dassDas von AlphaPPImd generierte Proteinkomplex-Konformationsmodell kommt der Referenzkristallstruktur näher.Die Genauigkeit war höher (RMS-Abweichung < 2 Å) und die Akzeptanz höher (DockQ ≥ 0,23).
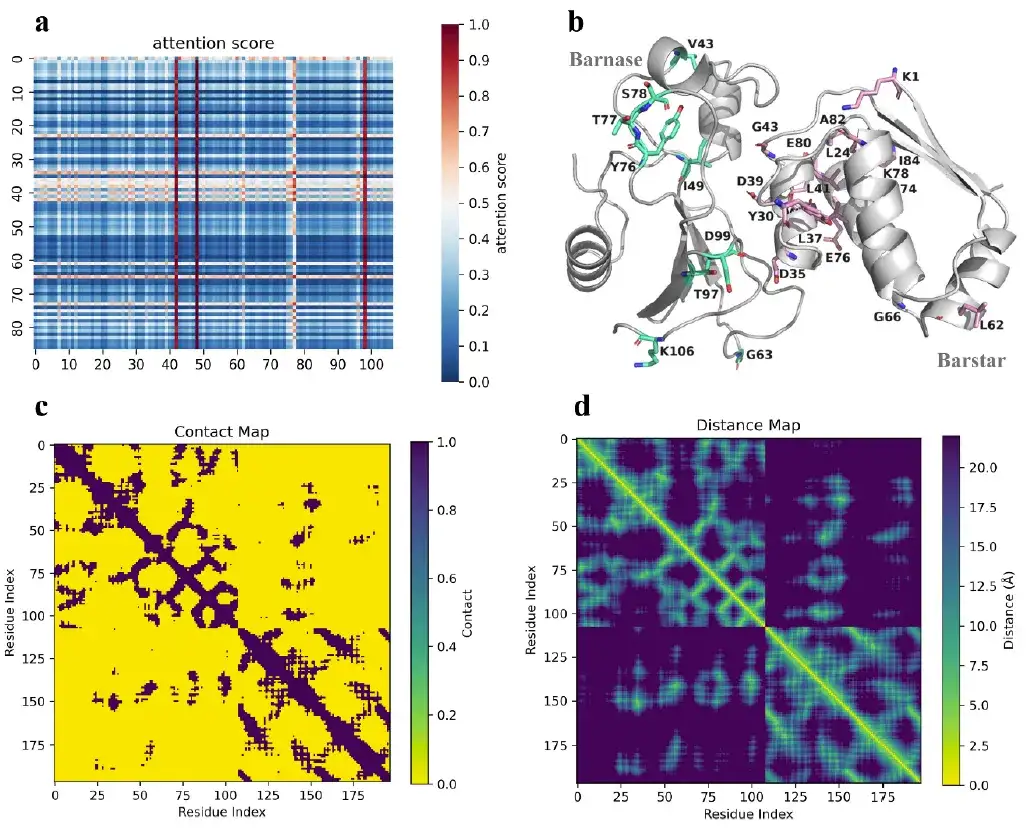
Darüber hinaus erfasst der Aufmerksamkeitsmechanismus von AlphaPPImd die Aufmerksamkeitsgewichte zwischen Schlüsselresten und bietet mechanistische Einblicke in die Protein-Protein-Bindung.
Studien haben gezeigt, dass die vom AlphaPPImd-Modell erfassten Schlüsselreste hauptsächlich an den Schnittstellen von Proteininteraktionen, Schleifen und Helices liegen, was bedeutet, dassDas tiefe generative Modell erfasste die Schlüsselreste, die die Dynamik und Konformation des Barnase-Barstar-Komplexes aus der MD-Trajektorie beeinflussen.Kann zur Ergänzung von MD-Ergebnissen verwendet werden. Die vom AlphaPPImd-Modell erfassten Schlüsselrückstände befinden sich inzwischen hauptsächlich in der Mdm2-p53-Interaktionsschnittstelle, was auch beweist, dass das Modell auf andere Protein-Protein-Komplexe erweitert werden kann.
KI-Proteinvorhersage: von AlphaFold Zu hundert Denkschulen
Bereits 2016, nachdem AlphaGo berühmt wurde, begann das DeepMind-Team mit der Erforschung des Proteinfaltungsproblems.
Beim 13. CASP (Critical Assessment of Protein Structure Prediction) Ende 2018 belegte AlphaFold unter 98 Teilnehmern den ersten Platz und sagte die Strukturen von 25 von 43 Proteinen korrekt voraus. Im Jahr 2020 wurde AlphaFold 2 eingeführt, das eine hochpräzise Vorhersage von Proteinmonomerstrukturen ermöglicht. Im Oktober 2021 veröffentlichte DeepMind ein Update namens AlphaFold-Multimer, das AlphaFold 2 erweitert und Komplexe aus mehreren Proteinen modellieren kann. Am 8. Mai 2024 überraschte AlphaFold 3 die Welt erneut und erweiterte den Vorhersageumfang von Proteinen auf eine breite Palette biologischer Moleküle.
Bereits bei der Markteinführung von AlphaFold 2 erklärte Shi Yigong, Mitglied der Chinesischen Akademie der Wissenschaften, den Medien: „Meiner Meinung nach ist dies der größte Beitrag der künstlichen Intelligenz zur Wissenschaft und zugleich einer der wichtigsten wissenschaftlichen Durchbrüche der Menschheit im 21. Jahrhundert. Es ist eine bemerkenswerte historische Errungenschaft in der wissenschaftlichen Erforschung der Natur durch die Menschheit.“
Am Beispiel von AlphaFold hat die durch KI ausgelöste industrielle Revolution im Bereich des Proteindesigns still und leise Einzug gehalten.
Im Jahr 2023Das weltweit erste KI-Proteingenerierungsmodell NewOrigin (chinesischer Name „Darwin“) wurde auf der World Manufacturing Conference offiziell vorgestellt.Es wird berichtet, dass das große Modell von NewOrigin auf einem bedingten Generierungsmechanismus basiert und mehrdimensionale Rückkopplungsmechanismen wie KI, Molekulardynamik, Quantencomputer und Nassexperimente kombiniert. Es kann Proteinsequenzen, Proteinfunktionen, Proteinwissensdarstellungen und andere modale Proteininhalte mit hoher Präzision generieren und mehrdimensionale Aufgaben wie Affinität, Stabilität, Aktivität und Expression erledigen, um die Anforderungen realer industrieller Anwendungen zu erfüllen.
Im Jahr 2022 veröffentlichten Biologen der University of Washington School of Medicine zwei Artikel in Science, in denen sie ihre wichtigsten Entdeckungen vorstellten. Die Forscher sagten:Mithilfe maschinellen Lernens können Proteinmoleküle in Sekundenschnelle erstellt werden.Früher dauerte dieser Zeitraum mehrere Monate. Die Schaffung von Proteinen, die in der Natur nicht vorkommen, wird bei der Impfstoffentwicklung helfen, die Forschung im Bereich der Krebsbehandlung beschleunigen, Instrumente zur Kohlenstoffabscheidung entwickeln, nachhaltige Biomaterialien entwickeln und vieles mehr.
Es besteht kein Zweifel, dass die KI-basierte Vorhersage von Proteinstrukturen uns helfen kann, Proteine und damit das Leben besser zu verstehen. Wissen und Verständnis allein reichen jedoch bei weitem nicht aus. In Zukunft werden Wissenschaftler KI zur Vorhersage von Proteinen einsetzen müssen, um praktische Probleme im medizinischen Bereich zu lösen, etwa um Proteine nach Bedarf zu modifizieren oder sogar Proteine von Grund auf neu zu entwickeln, die in der Natur nicht vorkommen. Der Weg vor uns ist lang und beschwerlich und wir freuen uns darauf, dass die KI bei der Erforschung der Biowissenschaften noch weitere Überraschungen mit sich bringt.








