Command Palette
Search for a command to run...
Die Forschungsgruppe Von He Yulian an Der Shanghai Jiaotong University Beschleunigt Das Katalysatordesign Und Extrahiert Automatisch Wissen Basierend Auf AutoML

Im täglichen Leben ist die „Katalyse“ eine der häufigsten chemischen Reaktionen. Das Wesentliche bei der Wein- und Essigherstellung ist beispielsweise der Prozess, bei dem die Stärke im Getreide unter Katalyse mikrobieller Enzyme in Alkohol und Essigsäure umgewandelt wird.
Um es etwas akademischer auszudrücken: Eine Substanz, die die Reaktionsgeschwindigkeit von Reaktanten in einer chemischen Reaktion ändern (entweder erhöhen oder verringern) kann, ohne das chemische Gleichgewicht zu verändern, und deren eigene Masse und chemische Eigenschaften sich vor und nach der chemischen Reaktion nicht ändern, wird als Katalysator bezeichnet.
In der chemischen Industrie sind alle Prozesse über 85% auf Katalysatoren angewiesen, um die Reaktionsgeschwindigkeit zu beschleunigen. Die Bedeutung der Entwicklung neuer und effizienter Katalysatoren für die gesamte Branche liegt auf der Hand.Eines der aufschlussreichsten Merkmale beim Verständnis und der Identifizierung des besten Katalysators ist die chemische Adsorptionsenergie E der Reaktanten auf der Katalysatoroberfläche.Anzeigen . Chemische Reaktionen sind von Natur aus komplex, was es schwierig macht, eindeutig zu bestimmen EAnzeigen Bei der Bestimmung der wesentlichen physikalischen Größen treten erhebliche Schwierigkeiten auf.
Kürzlich veröffentlichte die Forschungsgruppe von Assistenzprofessor Yulian He vom Joint Institute der Shanghai Jiao Tong University eine Forschungsarbeit mit dem Titel „Interpreting Chemisorption Strength with AutoML-based Feature Deletion Experiments“ in der international führenden Fachzeitschrift Proceedings of the National Academy of Sciences of the United States of America (PNAS).Ziel dieser Studie ist es, dieAnzeigen Es wird eine neue Methode zur automatischen Extraktion von Wissen aus einer Hochdurchsatz-Datenbank der Dichtefunktionaltheorie (DFT) vorgeschlagen, die auf Merkmalsentfernungsexperimenten auf Basis automatisierten maschinellen Lernens (AutoML) basiert.
Forschungshighlights:
* Experimente zur Merkmalsentfernung basierend auf automatisiertem maschinellem Lernen (AutoML), um automatisch Wissen aus Hochdurchsatz-DFT-Datenbanken (Dichtefunktionaltheorie) zu extrahieren
* Die Studie zeigt, dass die lokale geometrische Information der Adsorptionsstellen auf der Oberfläche von binären Legierungskatalysatoren einen wichtigen Einfluss auf die chemische Adsorptionsenergie E hatAnzeigen Die signifikanten Auswirkungen der AutoML-Feature-Entfernungsexperimente zeigen die Stabilität, Konsistenz und das Potenzial von
* Dieses Forschungsergebnis ist von großer Bedeutung für die Optimierung des Katalysatordesigns und hat erhebliche Auswirkungen auf die Methodik
Papieradresse:
https://www.pnas.org/doi/10.1073/pnas.2320232121
Folgen Sie dem offiziellen Konto und antworten Sie mit „Automatisches maschinelles Lernen“, um das vollständige PDF zu erhalten
Hochwertige Datensätze mit rigoroser Wissenschaft
Als Benchmark für diese Studie wurde ein mit der Dichtefunktionaltheorie berechneter Datensatz zur dissoziativen Chemisorptionsenergie ausgewählt. Die Datenqualität wurde durch Reproduktion der Adsorptionsenergien unter Verwendung desselben von Mamun et al. vorgeschlagenen DFT-Protokolls überprüft.
Diese Datenbank enthält DFT-berechnete E-Werte verschiedener Adsorbate auf binären Legierungsoberflächen.Anzeigen Die Adsorbate bestehen aus 37 verschiedenen Metallelementen. Die Forscher wählten dann chemische Adsorptionsreaktionen mit mehr als 10 Adsorbaten aus einem Datensatz mit 88.587 Einträgen aus und behielten nur fünf zweiatomige molekulare Adsorbentien (H2 , O2 , N2 , CO und NO), wie in der folgenden Tabelle gezeigt, mit insgesamt 8.418 Einträgen.

Der Hauptzweck der Beschränkung der Adsorbentien auf zweiatomige Moleküle besteht darin, die durch die Adsorbentstruktur verursachte Komplexität zu reduzieren und die Adsorbentbeschreibung zu vereinheitlichen, sodass sich das maschinelle Lernmodell auf das Oberflächenverhalten der betreffenden Legierung (d. h. des Katalysators) konzentrieren kann.
Methoden zur Wissensextraktion auf Basis von automatischem maschinellem Lernen (AutoML)
Bisher haben Forscher eher Methoden des maschinellen Lernens (ML) und insbesondere der erklärbaren künstlichen Intelligenz (XAI) verwendet, um neue Erkenntnisse über katalytische Reaktionen zu gewinnen. Angesichts der rasanten Entwicklung der KI-Technologie im Bereich der Chemie können die von XAI bereitgestellten Modelle und spezifischen Merkmalserklärungen jedoch möglicherweise nicht den Grad an Klarheit und Sicherheit erreichen, den Chemieforscher benötigen. daher,Diese Studie schlägt eine Alternative vor, nämlich einen automatisierten, auf maschinellem Lernen (AutoML) basierenden Ansatz zur Wissensextraktion.Wie unten gezeigt:
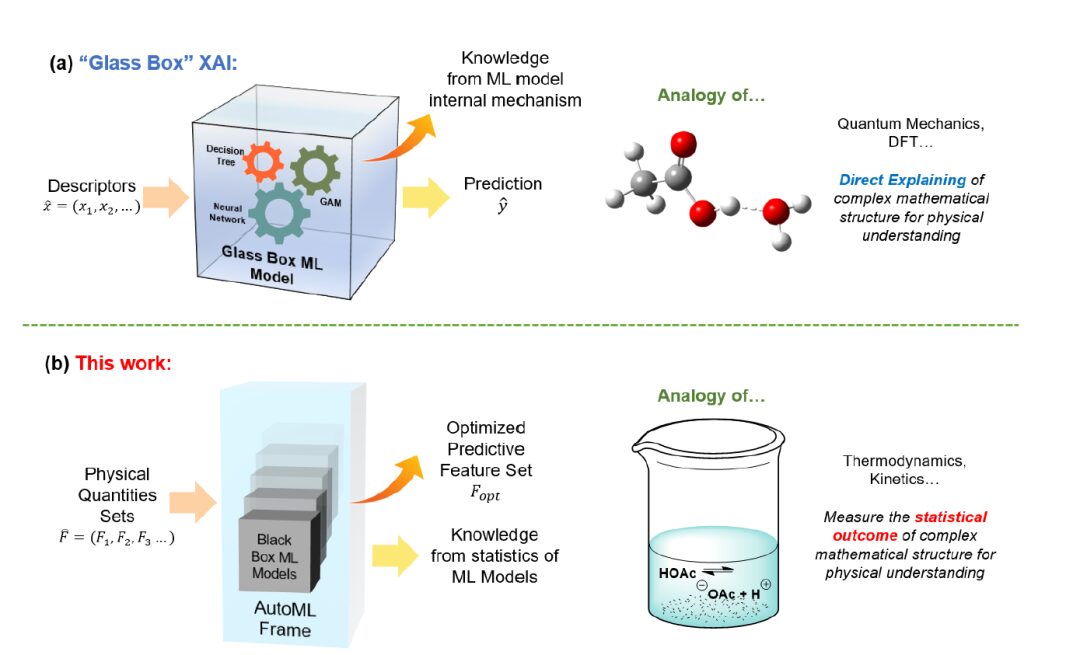
Anstatt tiefer in die Funktionsweise der Algorithmen des maschinellen Lernens einzutauchen, bündelten die Forscher viele vergleichbare Modelle des maschinellen Lernens für eine gemeinsame Analyse. Konkret bauten die Forscher ihre physikalischen Erkenntnisse auf einem einfachen, aber grundlegenden Prinzip auf – der Annahme, dass „kritische“ physikalische Größen die Vorhersagbarkeit physikalischer Modelle erheblich beeinflussen sollten; Daher würde das Entfernen dieser Mengen die Wirksamkeit des Modells verringern und umgekehrt.
erster Schritt, wird ein erster Benchmark-Feature-Satz (Ftotal) erstellt und validiert, um seine Beschreibungskraft sicherzustellen, und Modelle, die diesen Feature-Satz verwenden, sollten eine akzeptable Vorhersageleistung aufweisen.
Schritt 2, entfernen Sie intern korrelierte Merkmale aus Ftotal, um alle Änderungen in der Vorhersagbarkeit des Modells zu untersuchen.
Dieser Ansatz hat drei Vorteile:
1. Physikalische Erkenntnisse werden durch den Vergleich der Leistung verschiedener Funktionssätze gewonnen, wodurch physikalische Überlegungen explizit einbezogen werden. Durch sorgfältig konzipierte Versuchsaufbauten können Änderungen in der Vorhersagbarkeit mit physikalischen Hypothesen verknüpft werden.
2. Reduzieren Sie die Zufälligkeit des Modells, indem Sie die Statistiken vergleichbarer Modelle analysieren.
3. Dieser Ansatz vermeidet das Verständnis der detaillierten mathematischen Struktur von Algorithmen des maschinellen Lernens während des Wissensextraktionsprozesses und vermeidet so den Kompromiss zwischen Modellkomplexität und Interpretierbarkeit.
Forschungsergebnisse: Lokale geometrische Informationen von Adsorptionsstellen sind die entscheidende physikalische Größe
Durch maßgeschneiderte AutoML-basierte Experimente zur Feature-Entfernung,Diese Studie ergab, dass für binäre Legierungskatalysatoroberflächen die lokale geometrische Information der Adsorptionsstelle der Schlüsselfaktor ist, der die EAnzeigen Die entscheidende physikalische Größe sind nicht die intrinsischen elektronischen oder physikochemischen Eigenschaften des Legierungskatalysators.
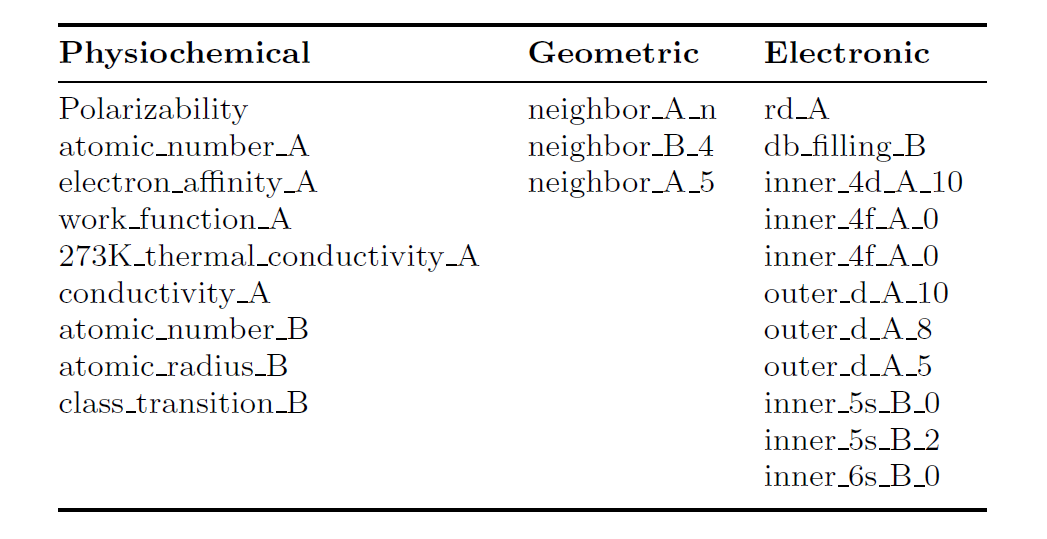
Insbesondere kombinierte die Studie Merkmalsentfernungsexperimente mit dem neuronalen Netzwerk-basierten Tool zur erklärbaren künstlichen Intelligenz (XAI) Instantiated Variable Selection (INVASE), um die Vorhersage von E zusammenzufassenAnzeigen Der beste Funktionssatz enthält 21 intrinsische, nicht per DFT berechnete intrinsische physikalische Größen F21. Mit diesem Funktionssatz wurde ein mittlerer absoluter Fehler (MAE) von 0,23 eV für ungefähr 8.400 chemische Adsorptionsreaktionen auf mehr als 1.600 Legierungsoberflächen erreicht.
Die folgende Tabelle zeigt die detaillierten Informationen zu F21, darunter 1 Adsorptionsmerkmal, 3 geometrische Merkmale, 7 physikochemischen Merkmale und 10 elektronischen Merkmalen.
Die Forscher wandten eine validierte Methode zur Merkmalsentfernung auf Ftotal an und ermittelten die relative Bedeutung der geometrischen, physikochemischen und elektronischen Merkmale von F21. Die Ergebnisse sind in der folgenden Abbildung dargestellt: Das Entfernen der elektronischen Merkmale aus F21 führt zu ΔMAE ≈ 0,04 eV, wodurch MAE = 0,30 eV wird, vergleichbar mit Ftotal.
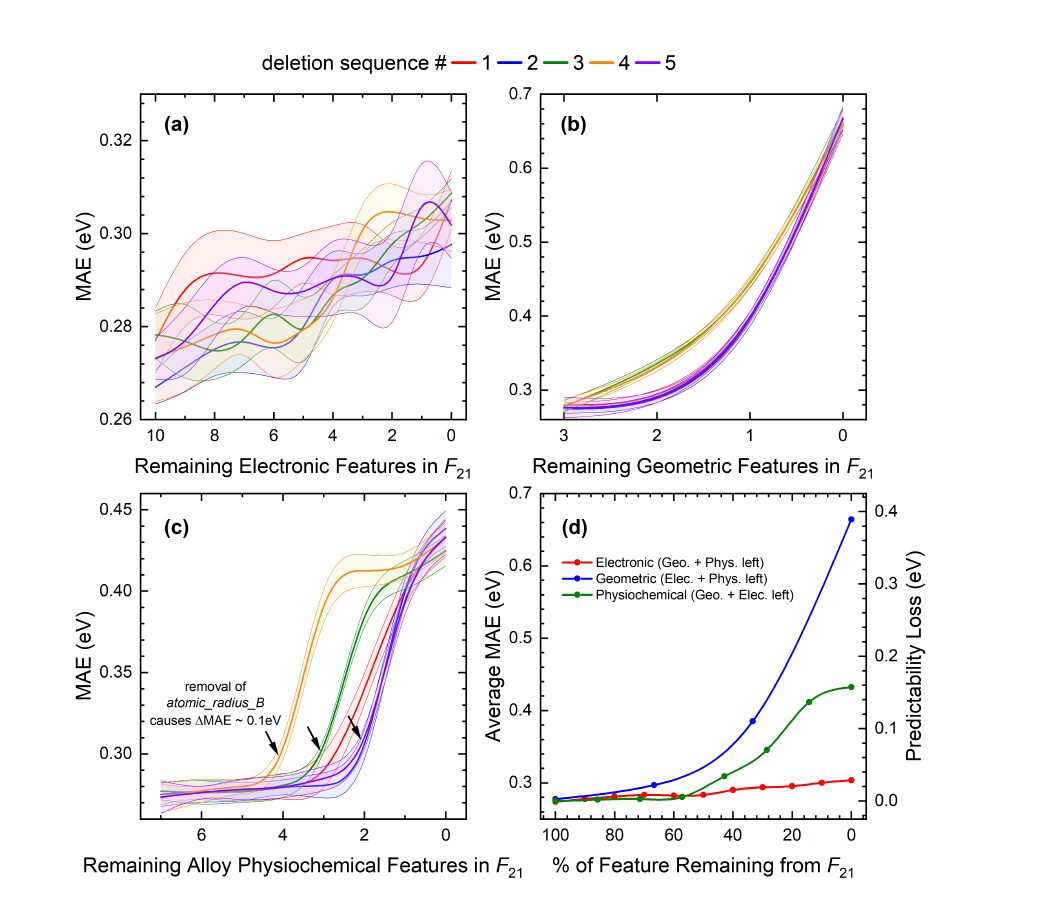
Ähnlich wie bei Ftotal werden zwar nur drei geometrische Merkmale ausgewählt, doch wie in Abbildung (b) oben gezeigt, spielen geometrische Informationen bei F21 mit einem ΔMAE von ungefähr 0,4 eV die wichtigste Rolle. Abbildung (c) oben zeigt, dass das Entfernen der physikochemischen Informationen zur Legierung aus F21 einen größeren Einfluss hat als die elektronischen Eigenschaften (ΔMAE ≈ 0,15 eV). Insbesondere stellten die Forscher fest, dass ein spezifisches Merkmal der Legierungskomponente B, der Atomradius B, besonders wichtig war. Unabhängig von der Reihenfolge der Löschung wurde ein ΔMAE von ungefähr 0,1 eV beobachtet, wenn der Atomradius B entfernt wurde. Die Bedeutung des Atomradius B könnte mit dem „Liganden“- oder „Spannungs“-Effekt in bimetallischen Nanokristallen zusammenhängen. Die Einführung eines sekundären Metalls B in die primäre Metallmatrix A kann erhebliche Änderungen des elektronischen Zustands und/oder der Gitterspannung (Kompression oder Spannung) hervorrufen und somit die chemische Adsorptionsstärke beeinflussen.
Wie in Abbildung (d) oben zusammengefasst, wird die relative Bedeutung von F21 wie folgt eingestuft: geometrisch > physikochemisch > elektronisch, was mit den Ergebnissen von Ftotal übereinstimmt.
Zusammenfassend zeigt diese Studie, dass die lokale geometrische Information der Adsorptionsstellen auf den Oberflächen von binären Legierungskatalysatoren einen signifikanten Einfluss auf die chemische Adsorptionsenergie E hat.Anzeigen Die Ergebnisse zeigen die erheblichen Auswirkungen der Merkmalsentfernung auf das Modell und demonstrieren die Stabilität, Konsistenz und das Potenzial von AutoML-basierten Merkmalsentfernungsexperimenten. Im Vergleich zu herkömmlichen interpretierbaren Modellen vermeidet diese Methode den Kompromiss zwischen Modellkomplexität und Interpretierbarkeit, verlagert die Quelle wissenschaftlicher Erkenntnisse von der Erläuterung des Modellverhaltens auf die Bewertung der Merkmalssatzleistung, minimiert die Auswirkungen menschlicher Eingriffe auf Schlussfolgerungen und extrahiert Erkenntnisse aus dem statistischen Verhalten der Ausgabe.
Diese neu vorgeschlagene, auf AutoML basierende Merkmalsanalysemethode ist ein leistungsstarkes und flexibles Tool, um die Bedeutung statistischer Merkmale in komplexen physikalischen Wissenschaften aufzudecken, auch über den Bereich der Katalyse hinaus.
Katalyse für eine effiziente Zukunft
Die Entwicklung neuer Katalysatoren ist der Schlüssel zur Lösung vieler Energie- und Umweltprobleme. Allerdings sind an vielen katalytischen Reaktionen einerseits komplexe Reaktionsmechanismen beteiligt, zu denen auch die Erzeugung und Umwandlung mehrerer Zwischenprodukte und Übergangszustände gehört. Diese Reaktionsmechanismen können durch viele Faktoren beeinflusst werden, wie etwa Lösungsmittel, Temperatur, Druck usw., was es sehr schwierig macht, die Leistung von Katalysatoren vorherzusagen und zu verstehen. Andererseits sind die Kosten für Versuch und Irrtum aufgrund der Komplexität und Unsicherheit der Katalysatorsynthese oft hoch. Bei herkömmlichen Methoden müssen möglicherweise viele verschiedene Materialien und Reaktionsbedingungen ausprobiert werden, was den Zeit- und Kostenaufwand für die Katalysatorentwicklung erhöht.
Um diese Herausforderungen zu bewältigen und die Designeffizienz und Leistung neuer Katalysatoren zu verbessern, müssen Techniken der künstlichen Intelligenz eingeführt werden. Künstliche Intelligenz kann mithilfe von Big Data und Algorithmen des maschinellen Lernens komplexe katalytische Reaktionsmechanismen analysieren und den Prozess der Katalysatorentwicklung und -optimierung beschleunigen. Zum Beispiel:
* Vorhersage und Design der Kristallstruktur:Mithilfe künstlicher Intelligenz kann die Kristallstruktur von Katalysatoren vorhergesagt und entworfen und so die katalytische Leistung verbessert werden. In der Vergangenheit suchten Wissenschaftler nach neuen Kristallstrukturen, indem sie bekannte Kristalle anpassten oder neue Elementkombinationen ausprobierten. Heute können Technologien wie Deep Learning große Mengen an Kristallstrukturdaten analysieren und Muster und Trends erkennen, die als Grundlage für die Katalysatorentwicklung dienen.
* Vorhersage und Optimierung chemischer Reaktionen:Künstliche Intelligenz kann dabei helfen, die Produkte und Reaktionswege chemischer Reaktionen vorherzusagen und die Reaktionsbedingungen zu optimieren, um den gewünschten katalytischen Effekt zu erzielen. Durch das Trainieren neuronaler Netzwerkmodelle können Wissenschaftler beispielsweise Vorhersagemodelle für Reaktionsmechanismen erstellen und diese als Leitfaden für die Versuchsplanung verwenden.
* Hochdurchsatz-Materialscreening:Künstliche Intelligenz kann den Hochdurchsatz-Screening-Prozess von Materialien beschleunigen und aus einer großen Anzahl von Materialkandidaten schnell Kandidaten mit potenziellen katalytischen Eigenschaften identifizieren.
* Intelligentes Experimentdesign und -optimierung:Künstliche Intelligenz kann beim Entwurf und der Optimierung experimenteller Protokolle helfen, um die Syntheseeffizienz und Leistung von Katalysatoren zu maximieren. Durch die Kombination von maschinellem Lernen und automatisierter Experimentiertechnologie kann eine intelligente Experimentierplattform erstellt werden, die experimentelle Prozesse automatisch ausführt und Anpassungen und Optimierungen auf der Grundlage von Echtzeitdaten vornimmt.
So demonstrierten Forscher der Universität Hokkaido im September 2023 beispielsweise einen extrapolierten Ansatz des maschinellen Lernens zur Entwicklung neuer Katalysatoren für die umgekehrte Wassergas-Shift-Technik mit mehreren Elementen. Mithilfe von 45 Katalysatoren als Ausgangsdatenpunkte und 44 Zyklen eines geschlossenen Entdeckungssystems (ML-Vorhersage + Experiment) testeten die Forscher experimentell insgesamt 300 Katalysatoren und identifizierten mehr als 100 Katalysatoren, die im Vergleich zu zuvor gemeldeten Hochleistungskatalysatoren eine höhere Aktivität aufwiesen.
Die Forschungsarbeit mit dem Titel „Beschleunigte Entdeckung multielementarer Reverse-Water-Gas-Shift-Katalysatoren mithilfe eines extrapolativen maschinellen Lernansatzes“ wurde in Nature Communications veröffentlicht.

In Zukunft dürfte künstliche Intelligenz die Design- und Syntheseeffizienz von Katalysatoren weiter verbessern, die Entdeckung und Anwendung neuer Katalysatoren beschleunigen und so die Entwicklung des Chemiebereichs vorantreiben.
Quellen:
1.http://www.sdqiying.com/cxinwenz/469/
2.https://www.zhihuiya.com/newknowledge/info_2859.html
3.https://www.ceshigo.com/article/11511.html
4.https://www.jiqizhixin.com/articles/2023-10-21-19








