Command Palette
Search for a command to run...
Die Tsinghua-Universität Ist Federführend Bei Der Veröffentlichung Des Uni-MOF-Modells, Das 630.000 Dreidimensionale Räumliche Konfigurationen Effektiv Identifiziert Und Die Adsorptionskapazität Von MOFs vorhersagt.

In der Industrie werden hochreine Gase in vielen Bereichen eingesetzt, beispielsweise in der Halbleiterherstellung, der Glasfaserproduktion, der wissenschaftlichen Forschung, der Medizin, im Umweltschutz und in der Energiebranche. In der Halbleiterindustrie sind hochreine Gase beispielsweise wichtige Rohstoffe für die Chipherstellung und wirken sich direkt auf die Leistung und Ausbeute integrierter Schaltkreise aus.
Die größte Herausforderung bei der Herstellung hochreiner Gase besteht in der Gastrennung. Zu den gängigen Methoden der Gastrennung zählen Kryotechnik (Destillationsprinzip), Adsorption (molekulare Polarität), Membranverfahren (Membranfiltration) usw. Unter diesen weisen Metall-organische Gerüstverbindungen (MOFs) aufgrund ihrer hochgeordneten Porenstruktur und einstellbaren Porengröße ein großes Anwendungspotenzial bei der Gasadsorption, -speicherung und -trennung auf.Manche sagen voraus, dass MOFs im 21. Jahrhundert eine ebenso große Bedeutung haben könnten wie Kunststoffe im 20. Jahrhundert.
Allerdings ist die genaue Vorhersage der Adsorptionskapazität von MOFs noch immer mit zahlreichen Herausforderungen verbunden. Um dieses Problem zu lösen, hat das Team von Professor Lu Diannan vom Institut für Chemieingenieurwesen der Universität Tsinghua in Zusammenarbeit mit Professor Wu Jianzhong von der University of California, Riverside, und dem Forscher Gao Zhifeng vom Beijing Institute of Scientific and Intelligent Technology kürzlich in Nature Communications eine neue Abhandlung mit dem Titel „Ein umfassender transformatorbasierter Ansatz für hochgenaue Vorhersagen der Gasadsorption in metallorganischen Gerüsten“ veröffentlicht.
Diese Studie schlägt ein maschinelles Lernmodell Uni-MOF zur Vorhersage des Adsorptionsverhaltens dreidimensionaler MOF-Materialien vor, das zur Vorhersage der Adsorptionsleistung nanoporöser Materialien für verschiedene Gase unter verschiedenen Arbeitsbedingungen verwendet wird.Dies ist ein wichtiger Durchbruch bei der Anwendung der Technologie des maschinellen Lernens im Bereich der Materialwissenschaften.
Forschungshighlights:
* Das Uni-MOF-Framework ist eine vielseitige Lösung zur Vorhersage der Gasadsorptionskapazität von MOFs unter verschiedenen Bedingungen
* Uni-MOF kann nicht nur die 3D-Struktur nanoporöser Materialien durch Vortraining erkennen und wiederherstellen, sondern berücksichtigt auch Betriebsbedingungen wie Temperatur, Druck und verschiedene Gasmoleküle, wodurch es sich sowohl für die wissenschaftliche Forschung als auch für praktische Anwendungen eignet
* Durch die Nutzung von Adsorptionsdaten anderer Gase kann Uni-MOF die Adsorptionsleistung unbekannter Gase genau vorhersagen

Papieradresse:
https://www.nature.com/articles/s41467-024-46276-x
Folgen Sie dem offiziellen Konto und antworten Sie mit „Adsorption“, um das vollständige PDF zu erhalten
Datensatz: vorhandene Datenbank + programmgenerierte Daten
In dieser Studie stammen die für das Vortraining verwendeten MOF/COF-Strukturen hauptsächlich aus zwei Aspekten – sie wurden aus aktuell verfügbaren Datenbanken gesammelt oder mithilfe entsprechender Programme generiert.
Derzeit gibt es eine große Anzahl von MOF/COF-Datenbanken, darunter die rechnerisch synthetisierte hMOFs50-Datenbank, das topologiebasierte Kristallkonstruktionsprogramm (ToBaCCo) MOFs und die experimentellen CoRE (Computational Ready Experiments) MOFs51, CoRE COFs52 und CCDC (Cambridge Crystallographic Data Center).
Darüber hinaus sind in der integrierten Online-Datenbank MOFXDB mehr als 168.000 MOF/COF-Strukturen verfügbar. Zusätzlich zur Untersuchung nanoporöser Materialien in der Materialbibliothek verwendeten die Forscher das Programm ToBaCCo.3.0, um mehr als 306.773 MOF-Strukturen zu generieren.
Für die nachfolgende Aufgabe, d. h. die Gasadsorption durch MOFs, sammelten die Forscher Daten aus Online-Quellen wie MOFXDB und erstellten einen Datensatz von mehr als 2,4 Millionen hMOFs für fünf Gase (CO2 , N2. CH4 , Kr, Xe) bei 273/298 K und 0,01–10 Pa und die Adsorptionsdaten von mehr als 460.000 CoRE MOFs für zwei Gase (Ar, N2) Adsorptionsdatensatz bei 77/87 K und 1–105 Pa.
Darüber hinaus führten die Forscher mit der Software RASPA54 Grand Canonical Monte Carlo (GCMC) 53-Simulationen durch und generierten dabei weitere 99.000 Gasadsorptionsdatensätze, darunter 50.000 Initialisierungszyklen und weitere 50.000 Zyklen für Adsorptionskapazitätsproben. Die Adsorptionsdaten wurden im Bereich von 150–300 K und 1 Pa–3 bar unter Berücksichtigung von sieben Gasmolekülen (CH4 、 CO2 , Ar, Kr, Xe, O2 , Er).
Modellrahmen: Vortraining + Feinabstimmung der Multitasking-Vorhersage
Das Uni-MOF-Framework umfasst ein Vortraining an dreidimensionalen nanoporösen Kristallen und eine Feinabstimmung für die Multitasking-Vorhersage in nachgelagerten Anwendungen.
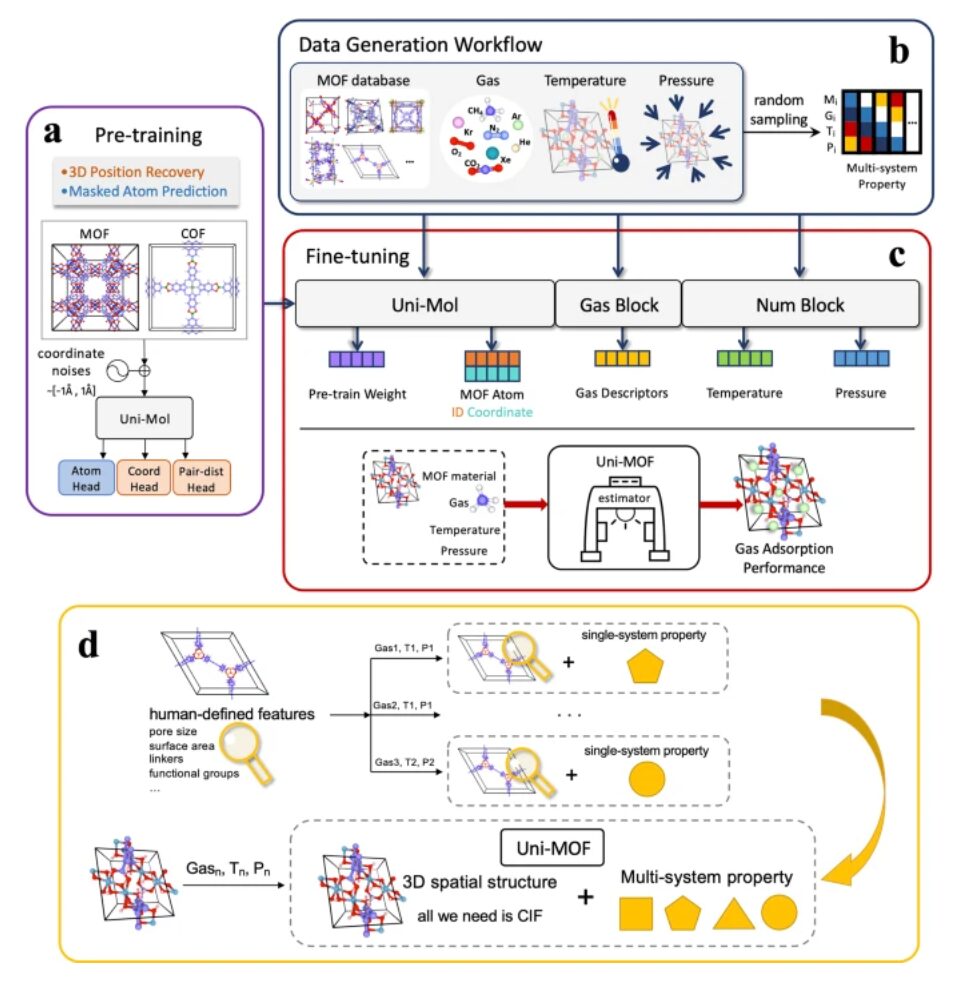
In der Vortrainingsphase des ModellsDie Forscher implementierten zwei Arten von Aufgaben, um die Modellleistung zu verbessern.
Die erste Art von Aufgabe besteht darin, die Art der maskierten Atome vorherzusagen, d. h. die Atomarten im maskierten Teil der Molekülstruktur zu identifizieren und vorherzusagen. Der zweite Aufgabentyp besteht darin, dreidimensionale Koordinatenwiederherstellungsaufgaben unter Rauschen durchzuführen. Die spezifische Operation besteht darin, gleichmäßiges Rauschen im Bereich von [-1Å, +1Å] auf den Atomkoordinaten von 15% einzuführen und dann die räumliche Positionskodierung basierend auf diesen beschädigten Koordinaten zu berechnen.
Diese beiden Aufgabentypen sollen die Widerstandsfähigkeit des Modells gegenüber Datenstörungen verbessern und so bei nachfolgenden Vorhersageaufgaben eine genauere Leistung bieten.
Während der FeinabstimmungsphaseDie Forscher verwendeten etwa 3 Millionen gekennzeichnete Datenpunkte, die MOFs und COFs unter einem breiten Spektrum von Adsorptionsbedingungen abdecken, um genaue Vorhersagen der Adsorptionskapazität zu erreichen.
Durch eine vielfältige Datenbank mit systemübergreifenden Zieldaten ist das fein abgestimmte Uni-MOF in der Lage, die Multisystem-Adsorptionsleistung von MOFs unter beliebigen Bedingungen, einschließlich unterschiedlicher Gase, Temperaturen und Drücke, vorherzusagen. Daher ist Uni-MOF ein einheitliches und einfach zu verwendendes Framework zur Vorhersage der Adsorptionseigenschaften von MOF-Adsorbentien.
Forschungsergebnisse: Uni-MOF-Gerüst bietet breite Anwendungsmöglichkeiten in der Materialwissenschaft
Zunächst validierten die Forscher die Vorhersagekraft von Uni-MOF.
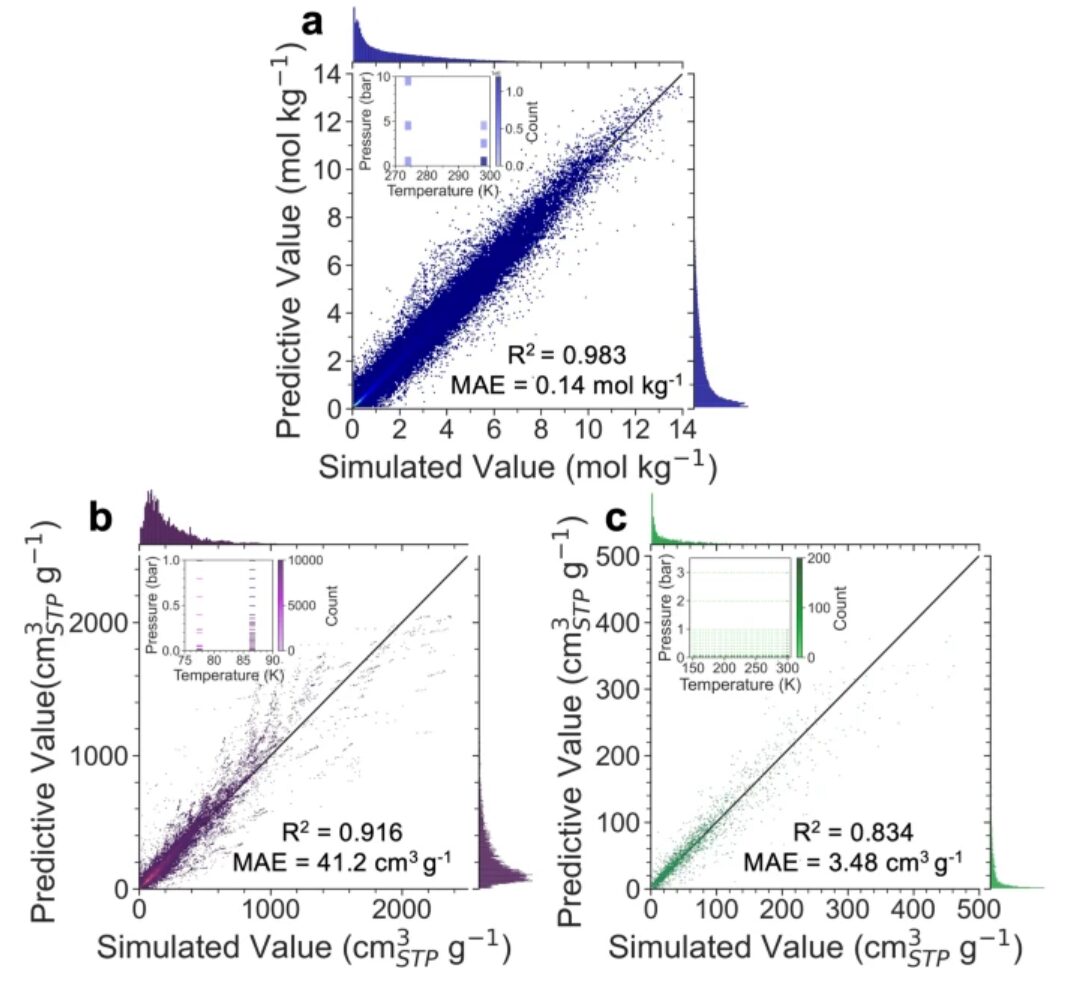
Die Vorhersageergebnisse zeigen, dass Uni-MOF bei Anwendung auf Datenbanken mit ausreichend Daten und relativ konzentrierten Betriebszuständen, wie z. B. hMOF_MOFX_DB und CoRE_MOFX_DB, eine sehr hohe Robustheit mit R²-Werten von 0,98 bzw. 0,92 aufweist. Auf dem weit verbreiteten Datensatz CoRE_MAP erreichte Uni-MOF eine Vorhersagegenauigkeit von 0,83, was immer noch eine hervorragende Vorhersagegenauigkeit erreichen kann und seine gute Generalisierungsfähigkeit demonstriert.
Zweitens verglichen die Forscher die von Uni-MOF vorhergesagten Ergebnisse mit experimentell gesammelten Ergebnissen.
Die Forscher stellten fest, dass das Uni-MOF-Framework in der Lage war, Hochleistungsadsorbentien ausschließlich auf der Grundlage vorhergesagter Adsorptionskapazitäten unter Niederdruckbedingungen genau zu screenen. Bemerkenswert ist, dass viele seiner unter Niederdruckbedingungen vorhergesagten Werte deutlich von den experimentellen Werten abweichen, insbesondere im Fall von Mg-dobdc und MOF-5. Dennoch zählt das Uni-MOF-Framework hinsichtlich der Vorhersagegenauigkeit unter vielen Materialien immer noch zu den besten und eignet sich daher zur Lösung technischer Herausforderungen.
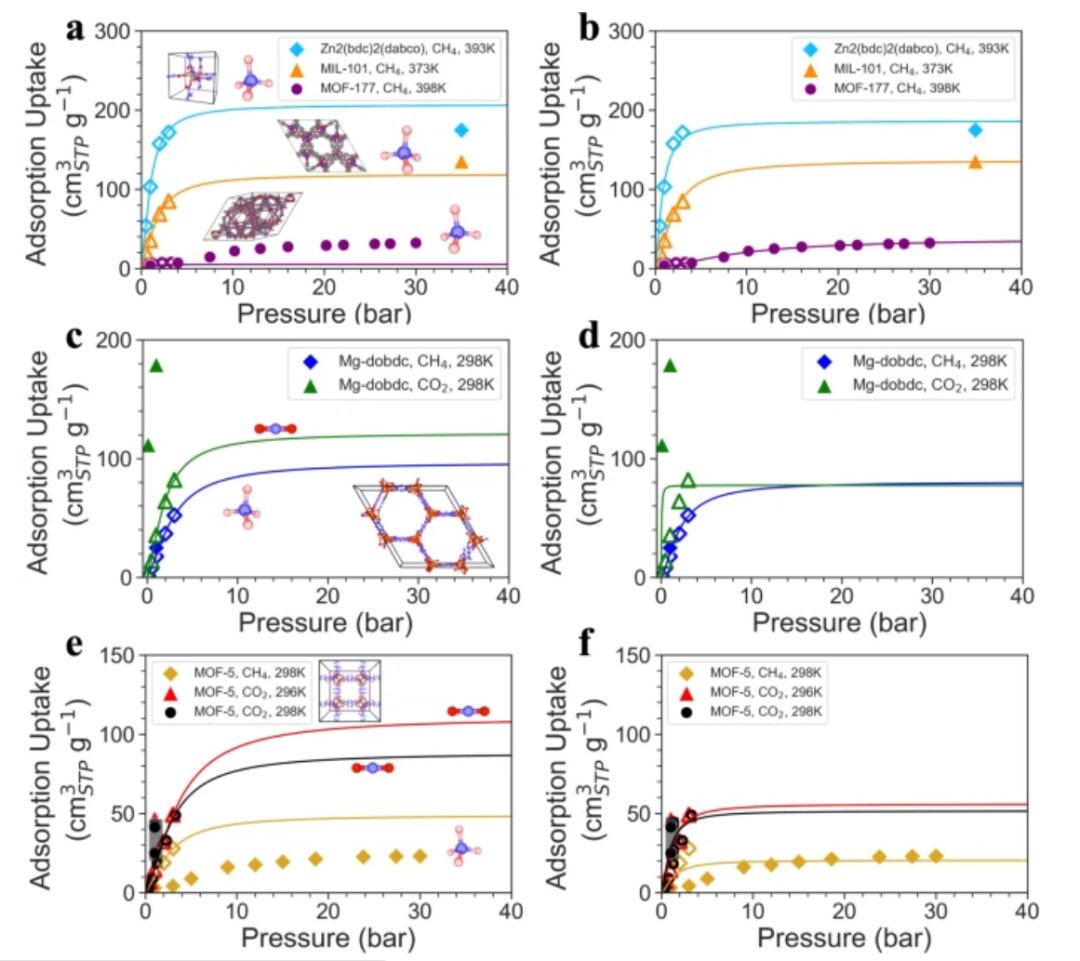
Jede Kurve stellt eine Langmuir-Anpassung dar
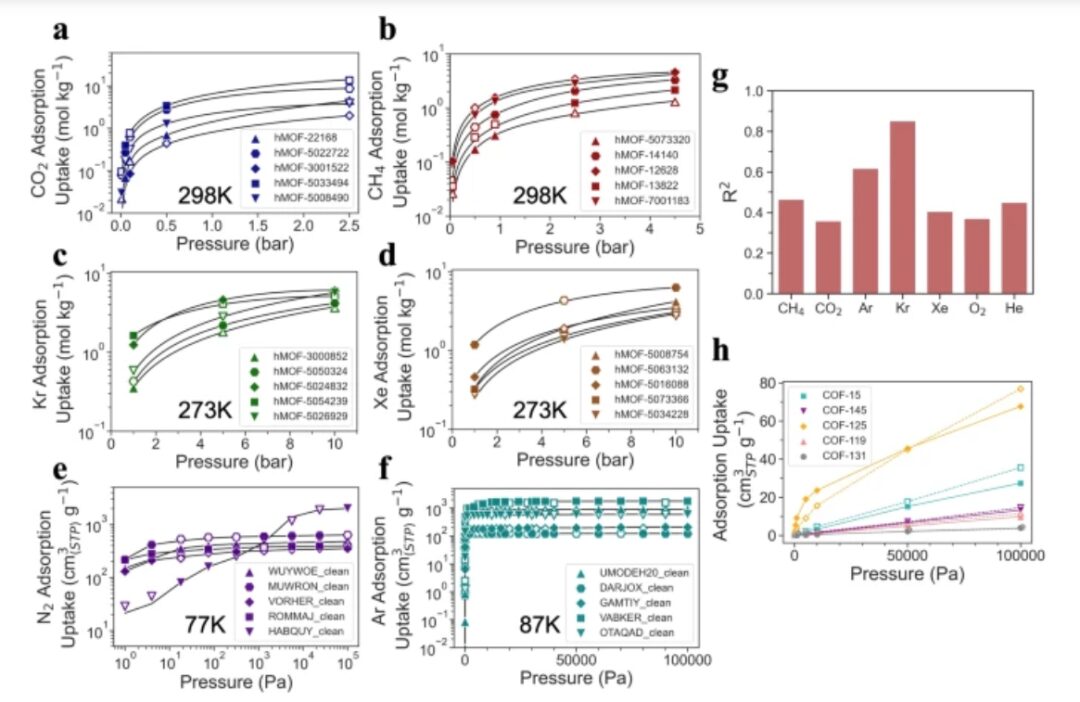
Drittens validierten die Forscher die Vorhersagekraft von Uni-MOF hinsichtlich systemübergreifender Eigenschaften.
Die experimentellen Ergebnisse zeigen, dass Uni-MOF die Adsorptionskapazität unbekannter Gase zuverlässig vorhersagen kann und eine hohe Vorhersagegenauigkeit (R²) von 0,85 für Krypton und eine Vorhersagegenauigkeit von über 0,35 für alle unbekannten Gase erreicht. Im Vergleich zu Einzelsystemaufgaben zeigt das Uni-MOF-Framework eine überlegene Leistung bei systemübergreifenden Datensätzen und kann die Adsorptionseigenschaften unbekannter Gase genau vorhersagen, was seine starke Vorhersagekraft und Universalität beweist.
Um außerdem die Fähigkeit des Modells zur Strukturerkennung zu bewerten, verwendeten die Forscher hMOF-5004238 als Beispiel, um die interatomaren Wechselwirkungen innerhalb der Materialstruktur zu analysieren.Beweisen Sie die Wirksamkeit von Uni-MOF bei der Identifizierung von mehr als 630.000 dreidimensionalen räumlichen Konfigurationen und ihren atomaren Verbindungen.Dies unterstreicht die Vielseitigkeit und die breiten Anwendungsmöglichkeiten des Modells.
Zusammenfassend ist das Uni-MOF-Framework eine vielseitige Vorhersageplattform für MOF-Materialien. Als Gasadsorptionsvorhersage für MOFs weist es eine hohe Genauigkeit bei der Vorhersage der Gasadsorption unter verschiedenen Betriebsbedingungen auf und findet breite Anwendung in der Materialwissenschaft. Noch wichtiger ist, dass Uni-MOF einen bedeutenden Durchbruch bei der Anwendung maschineller Lerntechniken im Bereich der Materialwissenschaften erzielt hat.
Entdeckung – Design – Optimierung, KI beschleunigt die Materialwissenschaft
Die Materialwissenschaft ist eine wichtige Disziplin im Zusammenhang mit der Entdeckung, Entwicklung und Herstellung neuer Materialien und spielt in vielen Bereichen eine äußerst wichtige Rolle. Von der Gesundheitsfürsorge bis zur Energiespeicherung, vom Umweltschutz bis zur Informationstechnologie sind Fortschritte in der Materialwissenschaft von entscheidender Bedeutung für die Lösung der vielfältigen Herausforderungen, vor denen die Gesellschaft heute steht.
Mit dem kontinuierlichen Fortschritt der Technologie befinden wir uns in einer Ära der Revolution in der Materialwissenschaft. Das Aufkommen neuer Materialien bietet der Menschheit neue Wege und Werkzeuge zur Lösung von Problemen. Wenn wir die Materialeigenschaften und -strukturen besser verstehen, können wir hoffentlich Materialien entwickeln, die leichter, stärker und energieeffizienter sind.
Künstliche Intelligenz kann die Entdeckung neuer Materialien beschleunigen, die Materialleistung verbessern und die Forschungs- und Entwicklungskosten senken. In den letzten Jahren hat es großes Anwendungspotenzial im Bereich der Materialwissenschaften gezeigt.
* Materialfindung und Design:
Künstliche Intelligenz kann den Entdeckungs- und Designprozess neuer Materialien durch effizientes Data Mining und Mustererkennung beschleunigen. Mithilfe von Algorithmen des maschinellen Lernens können beispielsweise die Struktur und Eigenschaften einer großen Anzahl bekannter Materialien analysiert und so neue Materialien mit bestimmten Eigenschaften vorhergesagt werden. Mit dieser Methode kann die Zeit für die Materialprüfung erheblich verkürzt und die Testkosten gesenkt werden.
Ende November 2023 veröffentlichte Google DeepMind einen Artikel in der Zeitschrift Nature, in dem es erklärte, es habe ein künstliches Intelligenz-Reinigungslernmodell namens Graph Networks for Materials Exploration (GNoME) für die Materialwissenschaft entwickelt und mithilfe dieses Modells und Hochdurchsatz-First-Principles-Berechnungen mehr als 380.000 thermodynamisch stabile kristalline Materialien gefunden, was „fast 800 Jahren Wissensansammlung menschlicher Wissenschaftler“ entspreche und die Forschungsgeschwindigkeit bei der Entdeckung neuer Materialien erheblich beschleunige.
* Vorhersage der Materialleistung:
Mithilfe künstlicher Intelligenz können effiziente Vorhersagemodelle erstellt werden, um die Leistung und das Verhalten von Materialien vorherzusagen. Diese Modelle können auf der Grundlage großer Mengen experimenteller Daten oder Simulationsergebnisse trainiert werden, um genaue Vorhersagen der Materialeigenschaften zu liefern. Beispielsweise können mithilfe von Algorithmen des maschinellen Lernens die mechanischen Eigenschaften, die thermischen Eigenschaften und die elektronische Struktur von Materialien vorhergesagt werden, was wichtige Referenzen für die Materialentwicklung und -anwendung liefert.
* Materialoptimierung und Design:
Künstliche Intelligenz kann die Leistung und Stabilität von Materialien verbessern, indem sie deren Struktur und Eigenschaften intelligent optimiert. Mithilfe von Reinforcement-Learning-Algorithmen lässt sich beispielsweise eine automatische Optimierung im Materialaufbereitungsprozess erreichen und so die Materialleistung maximieren.
* Materialprozesskontrolle und -überwachung:
Mithilfe künstlicher Intelligenz kann der Materialvorbereitungsprozess optimiert und eine intelligente Überwachung und Steuerung des Materialproduktionsprozesses realisiert werden. Mithilfe von Algorithmen des maschinellen Lernens können beispielsweise verschiedene Parameter und Bedingungen im Materialvorbereitungsprozess analysiert, der Prozessablauf optimiert und die Produktionseffizienz sowie die Materialqualität verbessert werden. Gleichzeitig ermöglicht die Technologie der künstlichen Intelligenz auch eine Echtzeitüberwachung und Frühwarnung des Materialproduktionsprozesses, wodurch potenzielle Probleme im Voraus erkannt und gelöst und Produktionsrisiken reduziert werden können.
Die Anwendung künstlicher Intelligenztechnologie im Bereich der Materialwissenschaften hat zu einer Reihe wichtiger Fortschritte geführt und neue Ideen und Methoden für die Entdeckung, Entwicklung, Optimierung und Herstellung von Materialien hervorgebracht. In Zukunft können Wissenschaftler mithilfe der KI-Technologie die Materialleistung besser vorhersagen, Molekülstrukturen simulieren, das Materialdesign optimieren, Materialeigenschaften untersuchen usw. und so den Fortschritt und die Innovation im Bereich der Materialwissenschaften kontinuierlich vorantreiben.
Quellen:
1.https://www.nature.com/articles/s41467-024-46276-x#Sec11
2.https://www.sohu.com/a/753459278_661314
3.https://www.tsinghua.edu.cn/info/1175/110086.htm








