Command Palette
Search for a command to run...
Unabhängige Forschung Und Entwicklung! Das Team Des Military Medical Research Institute Schlug MIDAS Vor, Das Für Die Mosaikintegration Von Einzelzell-Multi-Omics-Daten Verwendet Werden Kann
Wie wir alle wissen, sind Zellen die kleinsten Bausteine des Lebens. Der menschliche Körper enthält 40–60 Billionen Zellen, die die Grundlage unseres Wachstums und unserer Entwicklung bilden. Die Durchführung von Forschung auf Einzelzellebene ist für das genaue Verständnis von Zellwachstum und -entwicklung sowie für die Diagnose und Behandlung von Krankheiten von entscheidender Bedeutung.
In den letzten Jahren hat sich die Einzelzellsequenzierungstechnologie zu einem heißen Thema in der molekularbiologischen Forschung entwickelt. Die Branche hat eine große Menge an Einzelzellsequenzierungsdaten zu klinischen und grundlegenden Forschungsthemen wie Krankheit und Entwicklung generiert. Allerdings sind die riesigen Datenmengen aus unterschiedlichen Omics-Kombinationen, unterschiedlichen Sequenzierungstechnologien und unterschiedlichen Sequenzierungsproben so verstreut und vielfältig wie Mosaikfliesen auf dem Boden.Die Integration solch großer und komplexer Datenmengen und die Durchführung biomedizinischer Forschung ist für Wissenschaftler auf der ganzen Welt eine gemeinsame Herausforderung.
Um diese Herausforderung zu meistern, führten die Teams von Ying Xiaomin und Bo Xiaochen vom Militärmedizinischen Forschungsinstitut kürzlich Naturbiotechnologie Die Zeitschrift veröffentlichte ein Forschungspapier mit dem Titel „Mosaikintegration und Wissenstransfer von multimodalen Einzelzelldaten mit MIDAS“.In dieser Studie wurde das Computertool MIDAS für die Mosaikintegration von Einzelzell-Multimodal-Omics-Daten (scMulti-omics) (d. h., verschiedene Datensätze teilen sich nur einige Erkennungsmodalitäten) und den Wissenstransfer vorgeschlagen.Basierend auf selbstüberwachtem Lernen und informationstheoretischen Ansätzen haben wir zum ersten Mal die universellen Integrationsfunktionen von Einzelzell-Multi-Omics-Mosaikdaten wie modale Ausrichtung, Datenvervollständigung und Stapelkorrektur realisiert und so wichtige Originaltechnologien für die Konstruktion groß angelegter Multi-Omics-Zellkarten und die Realisierung groß angelegter Einzelzell-Multi-Omics-Analysen und Wissenstransfers bereitgestellt.
Forschungshighlights:
* Eigenständige Entwicklung eines neuen Algorithmus auf Basis generativer künstlicher Intelligenz, MIDAS
* Zum ersten Mal wurden die Integrationsfunktionen der Modalitätsausrichtung, Datenvervollständigung, Stapelkorrektur usw. von gängigen Einzelzell-Multi-Omics-Mosaikdaten realisiert
* Der neue Algorithmus ist von großer Bedeutung für die Aufklärung der Zellfunktionen und molekularen Regulationsmechanismen sowie für die Erforschung der Entstehung und Entwicklung von Krankheiten

Papieradresse:
https://www.nature.com/articles/s41587-023-02040-y
Folgen Sie dem offiziellen Konto und antworten Sie mit „Einzelzelle“, um das vollständige PDF zu erhalten
Datensatz: Mehrere Datensätze, mehrdimensionale Auswertungsleistung
Um die Vorteile des MIDAS-Modells aus verschiedenen Dimensionen zu vergleichen, wurden in dieser Studie mehrere Datensätze erstellt.
Erstens, um MIDAS mit modernsten Methoden zu vergleichen,In dieser Studie wurde die Leistung von MIDAS bei der trimodalen Integration mit vollständigen Modi (einer vereinfachten Form der Mosaikintegration) bewertet, einer Aufgabe, die das Forschungsteam „rechteckige Integration“ nannte. Das Team verwendete zwei veröffentlichte trimodale Einzelzell- PBMC Datensätze (DOGMA-seq und TEA-seq), wobei RNA, ADT und ATAC gleichzeitig in jeder Zelle gemessen werden, wodurch die Dogma-Full- und Teadog-Full-Datensätze erstellt werden. Hinweis: PBMC steht für periphere mononukleäre Blutzellen und wird häufig in wissenschaftlichen Forschungsaktivitäten im Bereich der Immunologie verwendet.
Zweitens, um die Leistung von MIDAS bei der Mosaikintegration zu bewerten,Basierend auf dem zuvor generierten rechteckigen Datensatz erstellte das Forschungsteam weitere 14 unvollständige Datensätze, von denen jeder durch Löschen mehrerer modaler Batchblöcke aus dem vollständigen modalen Datensatz generiert wurde.
Drittens, um die Wissenstransferfähigkeit von MIDAS zu untersuchen,Das Forschungsteam hat den Atlasdatensatz neu aufgeteilt in einen Referenzdatensatz, der für die Atlaskonstruktion verwendet wird, und einen Abfragedatensatz. Das Forschungsteam erhielt einen Referenzdatensatz mit dem Namen atlas-no_dogma, indem es DOGMA-seq aus dem Atlas entfernte.
Viertens soll die Anwendung von MIDAS in Einzelzelldatensätzen mit kontinuierlichen Zellzustandsänderungen untersucht werden.Das Forschungsteam erstellte einen menschlichen BMMC-Mosaik-Datensatz, indem es drei verschiedene Proben (ICA, ASAP und CITE) kombinierte, die aus öffentlicher scRNA-Sequenzierung (Einzelzell-RNA-Sequenzierung) gewonnen wurden.
Modellarchitektur: Tiefes generatives Modell MIDAS
MIDAS ist ein tiefes generatives Modell, das die gemeinsame Verteilung unvollständiger multimodaler Einzelzelldaten darstellt, darunter Messungen von transposasezugänglichem Chromatin (ATAC), RNA und antikörperabgeleiteten Tags (ADTs).
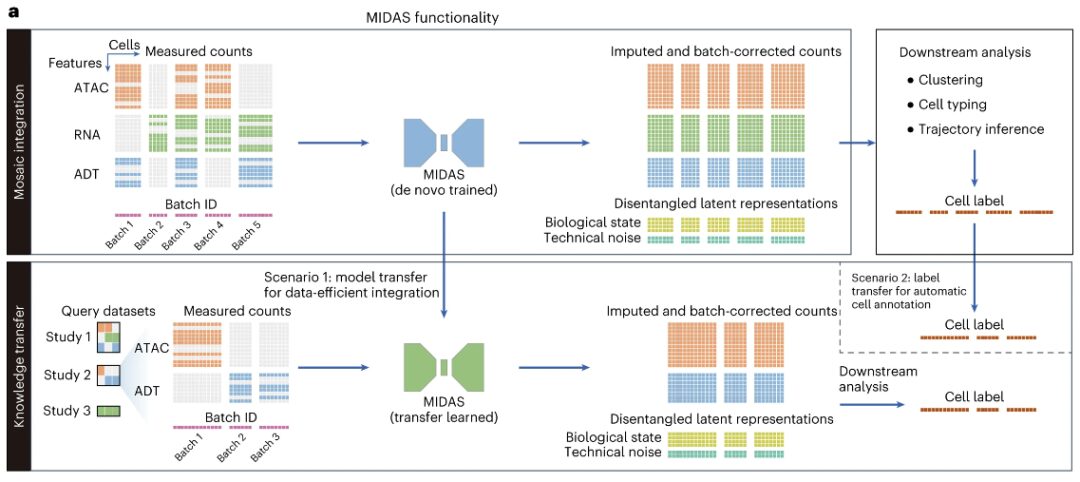
Insbesondere geht MIDAS davon aus, dass multimodale Messungen jeder Zelle über zwei modalitätsunabhängige und entkoppelte latente Variablen (biologischer Zustand und technisches Rauschen) basierend auf einem tiefen neuronalen Netzwerk generiert werden.Seine Eingabe umfasst eine Mosaik-Feature-Zellenzählungsmatrix, die aus verschiedenen Einzelzellproben (Chargen) und einem Vektor besteht, der die Zell-Chargen-ID darstellt.Diese Einzelzellproben können aus unterschiedlichen Experimenten stammen oder durch die Anwendung unterschiedlicher Sequenzierungstechnologien (wie scRNA-seq, CITE-seq, ASAP-seq und TEA-seq) erzeugt werden und können daher unterschiedliche technische Details, Modalitäten und Eigenschaften aufweisen.
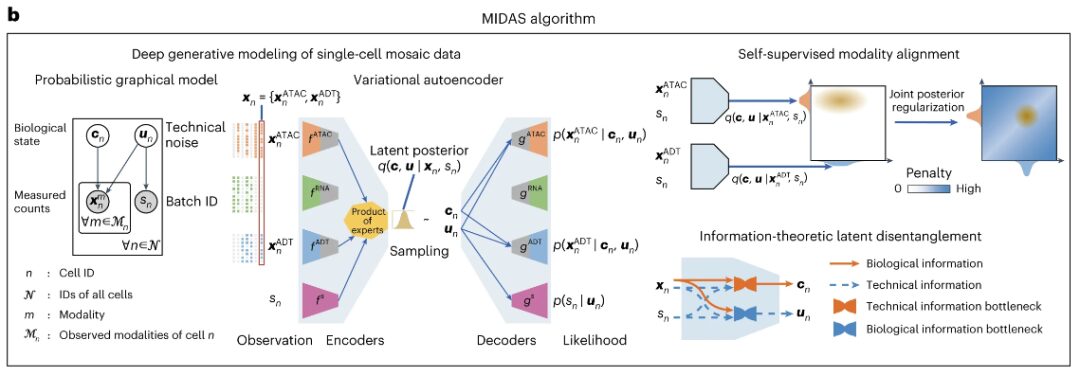
Zu den Ausgaben von MIDAS gehören biologische Zustands- und technische Rauschmatrizen sowie geschätzte und stapelkorrigierte Zählmatrizen, aus denen fehlende Modalitäten und Merkmale in den Eingabedaten interpoliert und Stapeleffekte entfernt werden.Diese Ausgaben können für nachgelagerte Analysen wie Clustering, Zelltypabgrenzung und Trajektorieninferenz verwendet werden.
MIDAS basiert auf der Variational Autoencoder (VAE)-Architektur und verfügt über ein modulares Encoder-Netzwerk und ein Decoder-Netzwerk. Erstere können Mosaik-Eingabedaten verarbeiten und latente Variablen ableiten, und letztere können latente Variablen verwenden, um den Generierungsprozess beobachteter Daten zu starten. MIDAS verwendet selbstüberwachtes Lernen, um verschiedene Modalitäten im latenten Raum auszurichten und so die kreuzmodale Inferenz in nachgelagerten Aufgaben wie Interpolation und Übersetzung zu verbessern. Darüber hinaus werden informationstheoretische Methoden angewendet, um den biologischen Zustand und das technische Rauschen zu entkoppeln und so eine weitere Batch-Korrektur zu erreichen.
Die Forscher kombinierten diese Elemente mit den Optimierungszielen dieser Studie und erreichten skalierbares Lernen und Inferenz von MIDAS durch stochastische Gradienten-Variations-Bayes-Methode (SGVB), was auch eine groß angelegte Mosaikintegration und Kartenerstellung von multimodalen Einzelzelldaten ermöglichte. Um das Wissen aus dem erstellten Atlas auf Abfragedatensätze mit unterschiedlichen Modalitätskombinationen zu übertragen, entwickelten die Forscher darüber hinaus Transferlern- und Querverweis-Mapping-Schemata für die Übertragung von Modellparametern bzw. Zellbeschriftungen.
Forschungsergebnisse: MIDAS ist vielseitig und effizient
Die Ergebnisse dieser Studie zeigen, dass MIDAS ein leistungsstarkes, vielseitiges und effizientes Tool zur multimodalen Integration einzelner Zellen ist.
Das Forschungsteam verglich die Leistung von MIDAS mit neun kürzlich veröffentlichten Methoden hinsichtlich der Beseitigung von Batch-Effekten und der Erhaltung biologischer Signale.
Die Ergebnisse zeigen, dassMIDAS eliminiert im Idealfall Batch-Effekte und bewahrt Zelltypinformationen in Dogma-Full- und Teadog-Full-Datensätzen, während die Leistung anderer Methoden etwas schlechter ist.Beispielsweise ließen sich die verschiedenen Batches bei BBKNN+average, MOFA+, PCA+WNN, Scanorama-embed+WNN und Scanorama-feat+WNN nicht gut mischen, und die von PCA+WNN und Scanorama-feat+WNN generierten Zellcluster stimmten größtenteils nicht mit den Zelltypen überein.
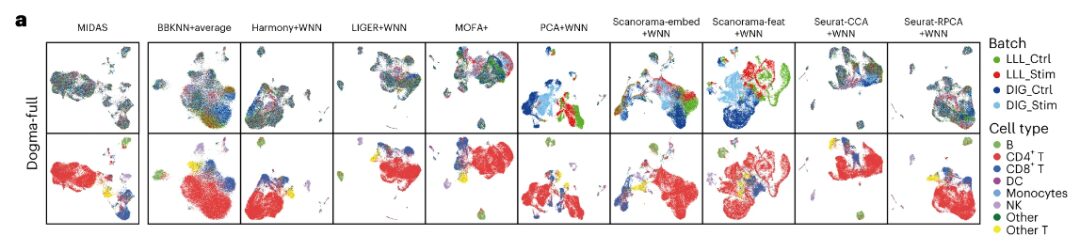
Erzielte Ergebnisse für die Auswertung und die nachfolgende Analyse
In Bezug auf die Stapelausrichtung – MIDAS ist in der Lage, Zellen aus verschiedenen Stapeln sehr gut auszurichten und sie konsistent mit Zelltypbezeichnungen zu gruppieren.Bei anderen Methoden lassen sich unterschiedliche Zellchargen nicht gut vermischen und es entstehen Cluster, die größtenteils nicht mit dem Zelltyp übereinstimmen. Der scIB-Benchmark zeigt, dass MIDAS bei verschiedenen Mosaikaufgaben eine stabile Leistung aufweist und sein Gesamtergebnis viel höher ist als bei anderen Methoden.
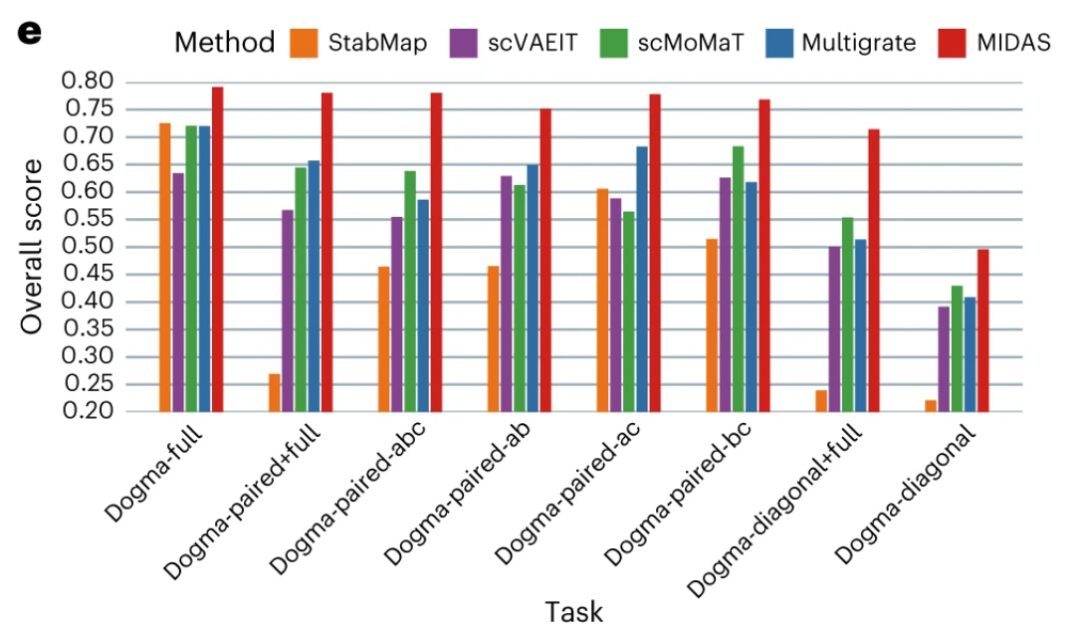
Qualitative und quantitative Leistungsbewertungsergebnisse
Im Hinblick auf die Fähigkeit zur Wissensübertragung haben die Forscher jeden Abfragedatensatz mit dem Referenzdatensatz abgeglichen und k-nächste Nachbarn verwendet (kNN) Algorithmus zum Übertragen von Zelltypbezeichnungen. Nach der Kartierung und Visualisierung der biologischen Zustände ist ersichtlich, dass die Ergebnisse der Querverweiskartierung verschiedener Abfragedatensätze mit den durch den Dogma-Volldatensatz erhaltenen Kartenintegrationsergebnissen übereinstimmen und in hohem Maße übereinstimmen. MIDAS ermöglicht eine robuste und genaue Tag-Übertragung, wodurch eine De-novo-Integration und nachgelagerte Analyse überflüssig wird.Daher kann MIDAS verwendet werden, um Wissen auf Atlasebene auf verschiedene Formen von Benutzerdatensätzen zu übertragen, ohne dass teure Neuschulungskosten oder komplexe nachgelagerte Analysen erforderlich sind.
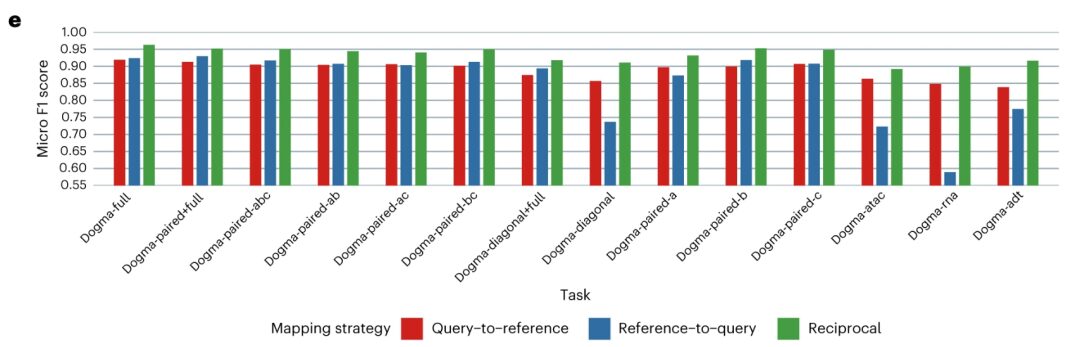
Zusammenfassend kann MIDAS durch die Modellierung des Prozesses zur Generierung von Mosaikdaten einzelner Zellen biologische Zustände und technisches Rauschen präzise von den Eingaben trennen und Modalitäten robust anpassen, um integrierte Analysen aus mehreren Quellen und heterogene Analysen zu unterstützen. MIDAS liefert genaue und robuste Ergebnisse und übertrifft andere Methoden bei der Durchführung verschiedener Mosaikintegrationsaufgaben.
Darüber hinaus überträgt MIDAS effizient und flexibel Wissen aus Referenzdatensätzen in Abfragedatensätze und erleichtert so die Verarbeitung neuer Multi-Omics-Daten. Mit hervorragender Dimensionsreduzierung und Batchkorrekturleistung unterstützt MIDAS eine genaue nachgelagerte Bioanalyse. MIDAS ermöglicht nicht nur die Clusterung und Zelltypidentifizierung von Mosaikdaten, sondern kann auch bei der pseudo-zeitlichen Analyse von Zellen mit sequenziellen Zuständen helfen, was besonders wertvoll ist, wenn keine RNAomics-Daten verfügbar sind. MIDAS ist in der Lage, heterogene Datensätze auszurichten und Zelltypen, sogar neue Typen, zu identifizieren, wenn Wissen zwischen verschiedenen Geweben übertragen wird.
Die Multi-Omics-Analyse einzelner Zellen schreitet weiter voran
So wie wir die Welt anhand eines Sandkorns erkennen können, können Wissenschaftler auch das Multiversum oder genauer gesagt die „Multi-Omics“ aus dem Inneren einer winzigen Zelle erkennen.
Zur Untersuchung des Genoms, Transkriptoms, Epigenoms und anderer Merkmale einzelner Zellen wird eine Reihe unterschiedlicher Techniken eingesetzt. Obwohl jede Technik für sich genommen aufschlussreich ist, liefert ihre kombinierte Analyse – die sogenannte Multi-Omics-Methode – ein umfassenderes Bild.Derzeit werden in der Zellbiologie und der translationalen Forschung, angetrieben durch die Einzelzell-Multiomik, erhebliche Fortschritte erzielt, doch die Datenintegration und -analyse bleibt für viele Wissenschaftler eine Herausforderung.
Auf dieser Grundlage ziehen neben den oben erwähnten Teams von Ying Xiaomin und Bo Xiaochen weitere Forschungsteams und Unternehmen nach und versuchen, effizientere und einfachere Wege der Datenverarbeitung zu erforschen.
Zum Beispiel,Analytische Methoden wie die Chromium-Einzelzellplattform von 10x Genomics werden ständig erweitert und ermöglichen die Bewertung mehrerer Zellmerkmale in unterschiedlichen Kombinationen.Einschließlich der Genexpression des gesamten Transkriptoms, der Proteinexpression und der vollständigen Paarung TCR und BCR-Sequenzierung, Antigenspezifität und offene Chromatinanalyse. Darunter Cell Ranger Die Lösung verwendet eine Reihe kostenloser und benutzerfreundlicher Analyse-Pipelines zur Analyse von Chromium-Einzelzelldaten, die Rohdaten verarbeiten und Ausrichtungen zum Zählen von Genen durchführen können. Darüber hinaus kann Cell Ranger auch in Cloud-Analyseplattformen integriert werden, um Daten zu überwachen, zu verwalten und zu verarbeiten.
Zum Beispiel,Am 2. Mai 2022 veröffentlichte die Forschungsgruppe von Gao Ge an der Peking-Universität/Changping-Labor in Nature Biotechnology eine Forschungsarbeit mit dem Titel „Multi-omics single-cell data integration and regulatory inference with graph-linked embedding“.Es wurde eine Deep-Learning-Methode namens GLUE vorgeschlagen, die auf einer Graphenkopplungsstrategie basiert und erstmals eine unbeaufsichtigte präzise Integration und regulatorische Inferenz von Millionen von Einzelzell-Multi-Omics-Daten ermöglichte.
Die kontinuierliche Weiterentwicklung dieser bioinformatischen Tools und Software wird Forschern dabei helfen, komplexe Multi-Omics-Datensätze zu interpretieren und die Entwicklung der Zellbiologie voranzutreiben. Sie ist von großer Bedeutung für die Aufklärung der Funktionen und molekularen Regulationsmechanismen von Zellen sowie für die Erforschung der Entstehung und Entwicklung von Krankheiten und kommt letztlich den Menschen zugute.
Quellen:
1.https://www.chinagut.cn/articles/ss/02bc1e86e3734acebff57395d6e044a6
2.https://m.ebiotrade.com/newsf/2023-10/20231023151001602.htm
3.https://news.bioon.com/article/e49a810955a1.html
4.https://m.thepaper.cn/newsDetail_forward_26137031








