Command Palette
Search for a command to run...
Die Forschungsgruppe Von Li Huashan Und Wang Biao an Der Sun Yat-sen-Universität Entwickelte Das SEN-Maschinenlernmodell, Um Materialeigenschaften Mit Hoher Genauigkeit Vorherzusagen

Inhalte im Überblick: Das Verständnis der globalen Kristallsymmetrie und die Analyse äquivarianter Informationen sind für die Vorhersage von Materialeigenschaften von entscheidender Bedeutung, aber vorhandene auf Faltungsnetzwerken basierende Algorithmen können diese Anforderungen nicht vollständig erfüllen. Um dieses Problem zu lösen, entwickelte die Forschungsgruppe unter der Leitung von Li Huashan und Wang Biao von der Sun Yat-sen-Universität ein maschinelles Lernmodell namens SEN, das die Interaktion zwischen inhärenter Kristallsymmetrie und Materialstrukturclustern genau wahrnimmt.
Schlüsselwörter: Deep Learning MP-Datenbank zur Vorhersage von Materialeigenschaften
Autor | Li Baozhu
Herausgeber | Sanyang
Die Kristallsymmetrie spielt eine Schlüsselrolle bei der Untersuchung der physikalischen Eigenschaften von Materialien, beim Verständnis der Kristallstruktur, beim Entwurf neuer Materialien und bei der Durchführung von Experimenten wie der Röntgenbeugung. Das Verständnis der Kristallsymmetrien kann dazu beitragen, Analysen zu vereinfachen, ein besseres Verständnis der Materialeigenschaften zu ermöglichen und Berechnungen der Materialleistung effizienter zu gestalten. Noch wichtiger ist, dass die Kristallsymmetrie auch die Ladungsverteilung, die optischen Eigenschaften, die magnetischen Eigenschaften und andere physikalische Eigenschaften des Materials direkt beeinflussen kann.
In den letzten Jahren hat maschinelles Lernen auf der Grundlage statistischer Mechanismen breite Anwendung gefunden. Aus der Perspektive des maschinellen Lernens kann Kristallsymmetrie als Invarianz und Äquivarianz von Materialien betrachtet werden. Allerdings fällt es den vorhandenen Algorithmen des maschinellen Lernens für Kristallmaterialien, die auf fortgeschrittenen Graphennetzwerken basieren, schwer, komplexe Materialinvarianz und -äquivarianz zu kodieren.
Obwohl der Stacked Capsule Autoencoder (SCAE) auch räumliche Symmetriemerkmale direkt aus den Originaldaten extrahieren kann, ist das traditionelle Kapselmodell immer noch nicht in der Lage, die Beziehung zwischen der Struktur und der Leistung komplexer Materialsysteme zu analysieren.
Angesichts der oben genannten HerausforderungenDie von Huashan Li und Biao Wang von der Sun Yat-sen-Universität geleitete Forschungsgruppe entwickelte ein maschinelles Lernmodell namens SEN (Symmetry-Enhanced Equivariance Network)., wodurch die schlechte Leistung von auf Faltung basierenden Algorithmen in hochsymmetrischen Raumgruppen überwunden und hochpräzise Vorhersagen von Materialeigenschaften in allen Raumgruppen erreicht werden. Die entsprechenden Ergebnisse wurden aktuell in „Nature Communications“ veröffentlicht.

Ähnliche Ergebnisse wurden in „Nature Communications“ veröffentlicht.
Holen Sie sich das Papier:
https://www.nature.com/articles/s41467-023-40756-2
01 Datensatz: 6.027 Kristallmaterialien in der MP-Datenbank
Die Forscher extrahierten die Eigenschaften kristalliner Materialien basierend auf dem Konzept der chemischen Umgebung und der Darstellungsmethode grafischer Modelle. Sie definierten seine chemische Umgebung durch die umgebenden Atome und Bindungen innerhalb des Grenzradius des Zielatoms und extrahierten den Atomtyp, die atomare Konnektivität und die Bindungslänge um jedes Atom aus dem Materials Project, einer Open-Source-Python-Datenbank für Materialanalysen.
Es wird berichtet, dassDie in dieser Studie zur Vorhersage der Bandlücke und der Bildungsenergie verwendeten Datensätze stammen aus der Materials Project-Datenbank. Die Datensätze der Bandlücke und der Bildungsenergie enthalten 6.027 (aufgeteilt in Trainingssatz, Validierungssatz und Testsatz im Verhältnis 8:1:1) bzw. 30.000 Materialien.Die beiden Datensätze bestehen aus 64 Elementen und decken die Elemente des Periodensystems mit Ausnahme der Edelgasgruppe, der Lanthanoiden, der Actinoiden und der radioaktiven Elemente ab.
Die Forscher verwendeten Dichtefunktionaltheorie-Berechnungen (DFT), um die Zusammensetzung von 6.027 kristallinen Materialien in der Materials Project-Datenbank vorherzusagen, und testeten die Leistung des SEN-Modells basierend auf den vorhergesagten Schlussfolgerungen.
Die in dieser Studie verwendeten Daten zur Kristallsymmetrie und chemischen Umgebung sind in der Zenodo-Datenbank verfügbar.
Besuchen Sie den Link:
https://doi.org/10.5281/zenodo.8142678
02 Modellarchitektur: einheitliches Training von 3 Modulen
Wie in der Abbildung unten gezeigt,Das SEN-Modell verwendet eine komplexe Deep-Learning-Architektur, die Module zur Merkmalsextraktion (FE), Symmetriewahrnehmung (SP) und Eigenschaftsvorhersage (PP) umfasst.
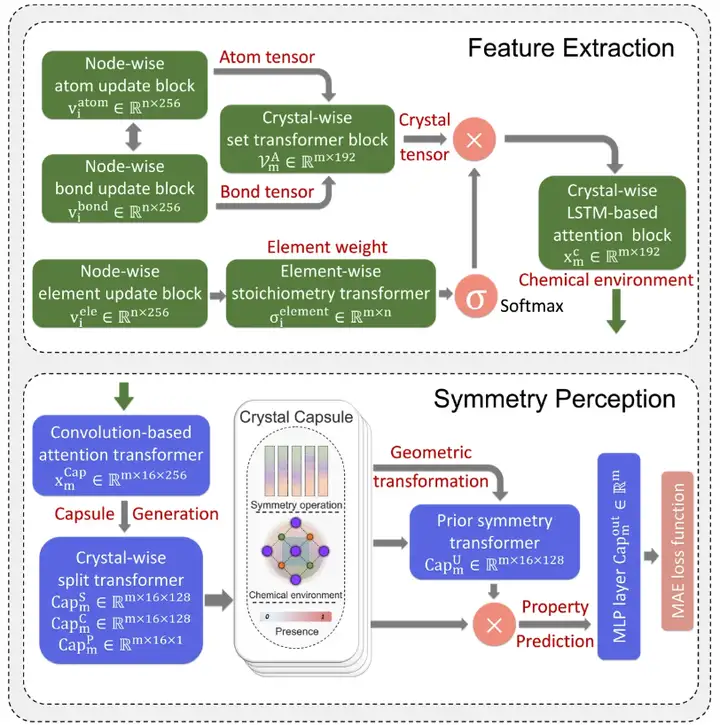
Die SEN-Architektur besteht aus Modulen zur Merkmalsextraktion, Symmetriewahrnehmung und Attributvorhersage.
In dieser Studie gelang es dem Forschungsteam, durch einheitliches Training von drei Modulen die Eigenschaften mehrerer Materialien genau vorherzusagen und die Wechselwirkung zwischen Atomen durch das SEN-Modell zu beschreiben.
Zunächst erfasst das Merkmalsextraktionsmodul die eingegebenen atomaren und chemischen Bindungsdaten, die die Informationen zu N-Atomen und M-Bindungen in der ursprünglichen Einheit des Zielmaterials enthalten. Schließlich wurde durch ein Hochdurchsatz-Screening-Verfahren ein Materialdatensatz erstellt, der Stöchiometrie, Kristallstruktur, Atominformationen und Bindungsinformationen umfasst.
Unter Verwendung des Materialdatensatzes als einzige Eingabedaten für das SEN-Modell berechneten die Forscher gleichzeitig den atomar-chemischen Umgebungsvektor VmA und den Elementgewichtsvektor VmE basierend auf den Strukturdaten und stöchiometrischen Daten.
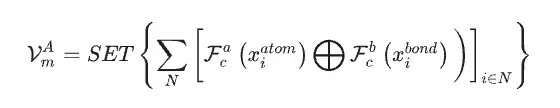
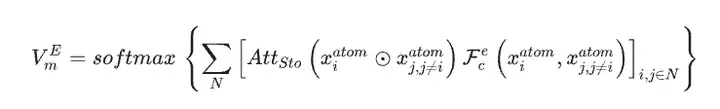
Nach der Aktivierung durch das mehrschichtige Perzeptron wird der Elementgewichtsvektor in einen Wahrscheinlichkeitsvektor des entsprechenden Atoms umgewandelt. Anschließend aktualisierten die Forscher alle Korrelationen auf atomarer Ebene durch elementweise Operationen zwischen atomaren chemischen Umgebungsvektoren und Elementgewichtsvektoren und erhielten so die chemische Umgebungsmatrix des Materials durch die LSTM-Aufmerksamkeitsschicht.
Zweitens wurde in dieser Studie der Kapselmechanismus auf innovative Weise zur Vorhersage von Materialeigenschaften angewendet. Durch das auf der Grundlage des Kapselmechanismus entwickelte Symmetriewahrnehmungsmodul wurde die materielle chemische Umgebung in eine materielle Kapsel umgewandelt, die aus Symmetrieoperatoren, einer gefalteten materiellen chemischen Umgebung und Existenzwerten besteht, um die Kristallsymmetrie wahrzunehmen und zu bewahren. Darüber hinaus können verschiedene Symmetriemuster auf die Kristallkapseln verallgemeinert werden, indem Symmetrieoperationen an der chemischen Umgebungsmatrix des Materials durchgeführt werden.
Und schließlich sagt das SEN-Modell im Hinblick auf die Eigenschaftsvorhersage die Zielmaterialeigenschaften durch eine MLP-basierte Abbildungsfunktion voraus.
03 SEN-Modell sagt Materialeigenschaften mit hoher Genauigkeit voraus
Schlussfolgerung 1: Das SEN-Modell nimmt atomare Interaktionsinformationen genau wahr
Um die Wirksamkeit des Merkmalsextraktionsmoduls zu überprüfen, trainierten die Forscher die Fähigkeit von SEN, die Bandlücke kristalliner Materialien vorherzusagen, bis der mittlere absolute Fehler (MAE) weniger als 0,15 eV betrug, und analysierten dann die vom Merkmalsextraktionsmodul generierten Zwischendaten zur chemischen Umgebung.
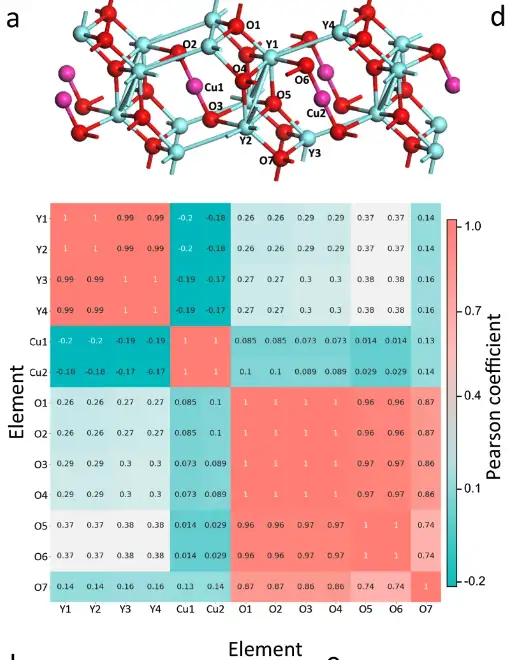
Atombasierte chemische Umweltkorrelationsanalyse
Insbesondere extrahierten die Forscher die chemische Umgebungsmatrix jedes Atoms in der Elementarzelle von Y4Cu2O7. Der Pearson-Koeffizient zwischen den Atommatrizen wurde berechnet, wodurch das oben gezeigte Diagramm der Korrelationsanalyse entstand. Die Pearson-Koeffizienten zwischen Atomen derselben Elementgruppe sind viel größer als die zwischen Atomen verschiedener Elementgruppen, sodass die drei Elementgruppen in Y4Cu2O7 klar unterschieden werden können.
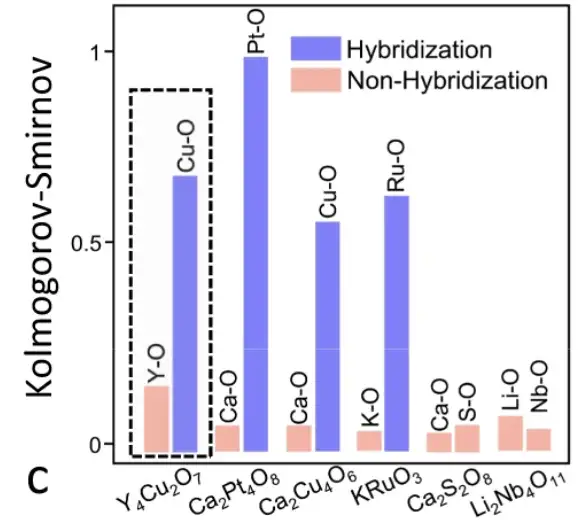
Die atomaren Korrelationen von sechs Materialien wurden vom SEN-Modell erlernt
Wie in der obigen Abbildung gezeigt, hat das SEN-Modell Informationen zur atomaren Interaktion gelernt und kodiert und das Hybridisierungsphänomen erfolgreich erkannt, das für die Vorhersage elektronischer Eigenschaften von großer Bedeutung ist.
Schlussfolgerung 2: Die Vorhersageleistung des SEN-Modells ist besser als die von MegNet
Um die Abbildung der chemischen Umgebung auf die Materialeigenschaften im SEN-Modell zu untersuchen, wählten die Forscher fünf Materialien aus der MP-Datenbank aus – Be(6)Ni(2), Sr(4)Ge(2)S(8), Li(2)V(2)F(12), CsAsF(6) und BaB(2)F(8) mit Bandlücken von 0 eV, 3,25 eV, 4,86 eV, 7,24 eV bzw. 10,12 eV.
Es wird beobachtet, dass eine starke Korrelation zwischen der Bandlücke und der PDF (Wahrscheinlichkeitsdichtefunktion) der chemischen Umgebung des Materials besteht, d. h., die PDF breitet sich allmählich aus, wenn die Bandlücke zunimmt. Die Projektion des gesamten Datensatzes von der chemischen Materialumgebung bis zur Bandlücke ist in der folgenden Abbildung dargestellt. Die 6.027 kristallinen Materialien sind gleichmäßig im Hauptmerkmalsraum verteilt, während die Änderung der Bandlücke im gesamten Raum kontinuierlich und monoton ist.
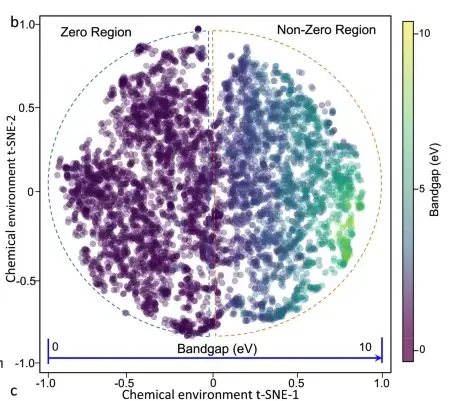
2D-t-SNE-Diagramm von 6027 Materialien. Die Farbe des Kreises gibt den Bandlückenwert an.
Um zu überprüfen, ob die vom maschinellen Lernmodell erlernten Merkmals-Attribut-Beziehungen mit grundlegenden physikalischen Prinzipien übereinstimmen, erstellten die Forscher eine 2D-t-SNE-Karte der chemischen Umgebung des Ca-OX-Materials und untersuchten verschiedene Materialeigenschaften (Zusammensetzung, Punktgruppe, Spinpolarisation usw.). Sie fanden schließlich heraus, dass die Materialbandlücke von komplexen Materialeigenschaften abhängt und nicht einfach durch einen Schlüsselfaktor vorhergesagt werden kann.
Dennoch erzielt das SEN-Modell erhebliche Verbesserungen bei der Bandlückenvorhersage.Das SEN-Modell erreicht einen mittleren quadratischen Fehler (MAE) von 0,25 eV bei der Vorhersage der Bandlücke von Materialien im Testdatensatz. Dies stellt eine erhebliche Verbesserung gegenüber dem MAE dar, der von Modellen mit MLP-, DenseNet-, TFN-, SE(3)- und EGNN-Modulen im Testdatensatz erzielt wird.
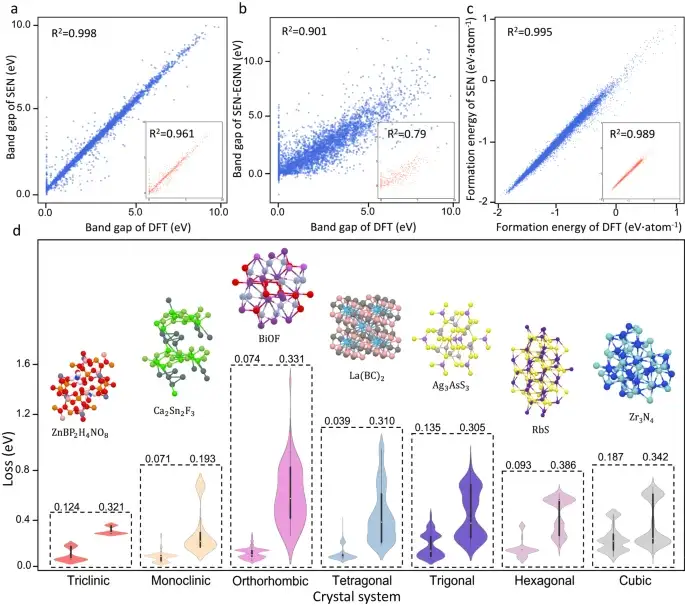
Vorhersage der Eigenschaften von Kristallmaterialien mit unterschiedlicher Symmetrie
Wie in Abbildung d oben gezeigt, verglichen die Forscher die Vorhersagequalität des SEN-Modells und des MegNet21-Modells (allgemeines Materialnetzwerkmodell) für verschiedene Kristallsysteme und verdeutlichten dabei den erheblichen Einfluss der Symmetriewahrnehmung auf die Vorhersage von Materialeigenschaften.Aus dem Fehlerverteilungsdiagramm ist ersichtlich, dass die Vorhersageleistung des SEN-Modells in allen Kristallsystemen besser ist als die von MegNet.
Darüber hinaus reduziert das SEN-Modell die effektive charakteristische Dimension erheblich, indem es die vollständige Kristallsymmetrie erfasst. Dieser Feature-Clearing-Prozess lindert das Überanpassungsproblem und verbessert die Zuordnung von Materialmerkmalen zu Eigenschaften.
Das Papier zeigt, dassDie mittleren absoluten Fehler der vom SEN-Modell vorhergesagten Bandlücke und Bildungsenergie sind ungefähr 22,9% bzw. 38,3% niedriger als die von gängigen Modellen des maschinellen Lernens.
04 KI fördert die Transformation und Entwicklung der Werkstoffindustrie
Design, Forschung und Entwicklung neuer Materialien sowie die Verbesserung der Materialeigenschaften sind seit langem eine der treibenden Kräfte für den wissenschaftlichen und technologischen Fortschritt und spielen in vielen Bereichen wie Elektronik, Energie, Medizin, Luft- und Raumfahrt usw. eine wichtige Rolle. Der traditionelle Prozess der Materialforschung und -entwicklung erfordert jedoch oft eine große Anzahl von Experimenten, um die Leistung kontinuierlich zu korrigieren und die Machbarkeit zu verbessern. Dieser Prozess ist langwierig und erfordert enorme personelle und finanzielle Ressourcen.
Mit der beschleunigten Anwendung von KI hat die KI für die Wissenschaft immer mehr Aufmerksamkeit erhalten und ihre Kombination mit Materialien ist für immer mehr Wissenschaftler und Unternehmen zu einer neuen Forschungsrichtung geworden. Einerseits kann KI große Datenmengen analysieren und Simulationsvorhersagen durchführen, wodurch die Entdeckung neuer Materialien beschleunigt und ihre Leistung optimiert wird. Andererseits ist die Materialwissenschaft auch zu einem wichtigen Standbein für zentrale KI-Technologien wie maschinelles Lernen, Verarbeitung natürlicher Sprache und Hochleistungsrechnen geworden.
Man kann sagen, dass KI still und leise das Design und die Anwendung neuer Materialien verändert. Durch die kontinuierliche Iteration leistungsfähigerer KI-Modelle und die Aktualisierung und Erweiterung von Materialdatenbanken im Rahmen der gemeinsamen Datennutzung wird KI in Zukunft die Entstehung neuer Materialien weiter fördern.








