Command Palette
Search for a command to run...
DeepMind Nutzt Unüberwachtes Lernen Zur Entwicklung Von AlphaMissense Und Sagt 71 Millionen Genmutationen Voraus

Das menschliche Genom besteht aus insgesamt 3,16 Milliarden Basenpaaren, die ständig einer Replikation, Transkription und Translation unterliegen und jederzeit der Gefahr von Fehlern und Mutationen ausgesetzt sind.
Missense-Mutationen sind eine häufige Form der Genmutation, aber bisher wurde beim Menschen nur ein kleiner Teil davon beobachtet und nur 0,1% können interpretiert werden.
Die genaue Vorhersage der Auswirkungen von Missense-Mutationen spielt eine wichtige Rolle bei der Erforschung und Prävention seltener Krankheiten und genetischer Erkrankungen. Dieses Mal griff DeepMind erneut ein.
Autor | Xuecai
Herausgeber | Drei Schafe, Eisenturm
Dieser Artikel wurde zuerst auf der öffentlichen HyperAI WeChat-Plattform veröffentlicht~
Das menschliche Genom verfügt insgesamt über 3,16 Milliarden Basenpaare. Diese Basenpaare werden täglich repliziert, transkribiert und translational und werden letztendlich als Proteine exprimiert, die die täglichen physiologischen Aktivitäten des Menschen regulieren.
Bei einer so großen Arbeitsbelastung wäre es selbst für den empfindlichen menschlichen Körper schwierig, perfekte Fehler zu erzielen.Wenn Sie nicht aufpassen, kann es zu einer Fehlausrichtung der Basenpaare kommen, was zu Genmutationen und im Laufe der Zeit sogar zu Krebs führen kann.
Die Missense-Mutation ist eine häufige Form der Genmutation.Durch Basenmutationen in der DNA verändern sich die übersetzten Aminosäuren, was letztendlich zur Zerstörung der gesamten Proteinfunktion führt.
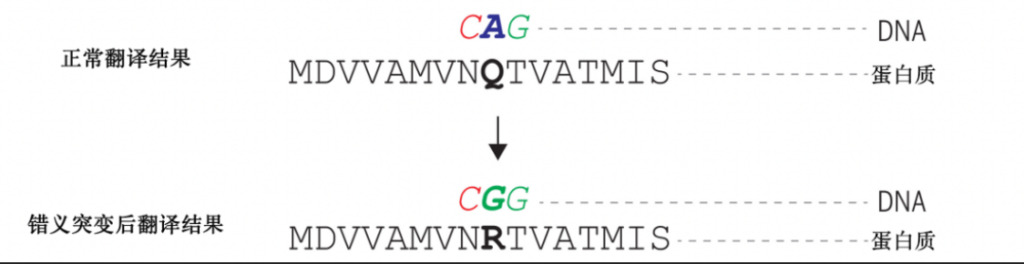
Abbildung 1: Schematische Darstellung der Missense-Mutation.Aufgrund der Mutation von Adeninnukleotiden zu Guaninnukleotiden in der DNA,Die Aminosäure, die von Glutamin in Serin umgewandelt wurde
Derzeit wurden beim Menschen mehr als 4 Millionen Missense-Mutationen beobachtet, aber nur die Missense-Mutationen von 2% können als pathogene oder gutartige Mutationen klassifiziert werden.
Durch die genaue Vorhersage der Auswirkungen von Missense-Mutationen können wir unser Verständnis seltener Krankheiten vertiefen und potenziellen genetischen Krankheiten vorbeugen und sie behandeln.Obwohl die Multiplexanalyse von Varianteneffekten (MAVEs) Proteinmutationen systematisch analysieren und ihre klinischen Auswirkungen genau vorhersagen kann,Allerdings erfordert diese Methode einen hohen Personal- und Materialaufwand.Es ist schwierig, eine umfassende Analyse aller möglichen Missense-Mutationen durchzuführen.
Zu diesem Zweck analysierte DeepMind die Gesamtstruktur des Proteins mittels AlphaFold.Wir haben AlphaMissense durch die Kombination von schwachem Label-Learning und unüberwachtem Lernen entwickelt.Die Folgen von Missense-Mutationen wurden systematisch analysiert. AlphaMissense wurde mithilfe des ClinVar-Datensatzes validiert.Die Vorhersagegenauigkeit erreichte 90%.
Dann,AlphaMissense prognostiziert 71 Millionen mögliche Missense-Mutationen beim Menschen, wobei 32% eine pathogene Mutation und 57% eine gutartige Mutation sein könnte.Diese Ergebnisse werden die Entwicklung von Disziplinen wie Molekularbiologie, Genomik und klinischer Medizin erheblich vorantreiben.Dieses Ergebnis wurde in „Science“ veröffentlicht.
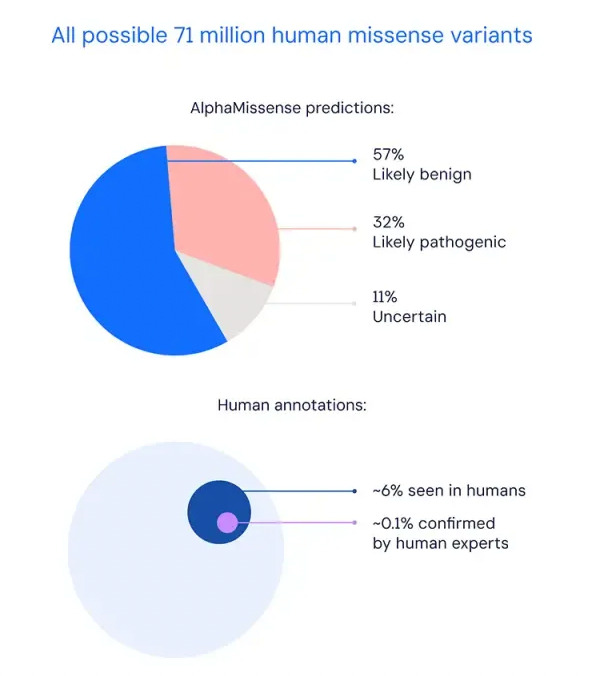
Abbildung 2: AlphaMissenses Vorhersageergebnisse für 71 Millionen Missense-Mutationen (oben) und die aktuell vom Menschen beobachteten und bestätigten Ergebnisse (unten)

Ähnliche Ergebnisse wurden in „Science“ veröffentlicht
Link zum Artikel:
https://www.science.org/doi/10.1126/science.adg7492
Experimentelle Verfahren
AlphaMissense:AlphaFold + Feinabstimmung
Wenn eine Aminosäuresequenz in AlphaMissense eingegeben wird, sagt es die Pathogenität jeder Aminosäureänderung in der Sequenz voraus. Die Implementierung von AlphaMissense ist AlphaFold sehr ähnlich, mit nur geringfügigen Anpassungen an der Architektur.
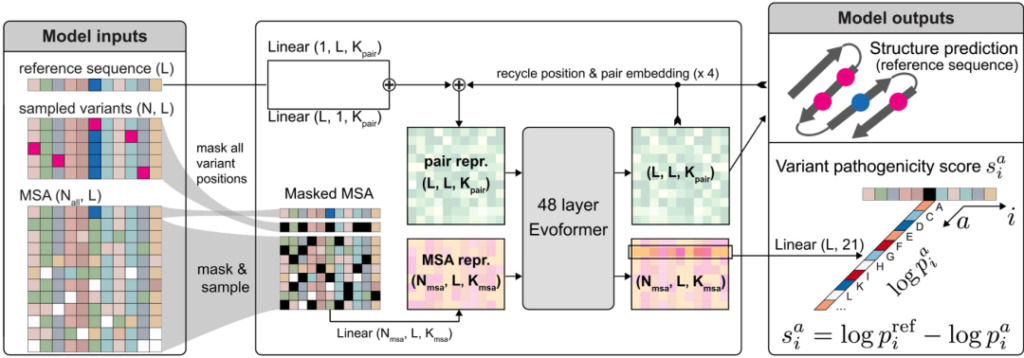
Abbildung 3: AlphaMissense-Strukturdiagramm
Die Trainingsdaten von AlphaMissense stammen aus einer Vielzahl von Quellen, hauptsächlich jedoch von Menschen und nichtmenschlichen Primaten.Darunter befinden sich 1.248.533 gutartige Missense-Mutationen, die vom Menschen stammen, und pathogene Missense-Mutationen werden aus 65.314.044 Mutationen extrahiert, die auftreten können, aber noch nicht beobachtet wurden.
Das Training von AlphaMissense besteht aus zwei Schritten. Zunächst muss AlphaMissense wie AlphaFold die zufällig maskierten Aminosäuren in mehreren Sequenzalignments vorhersagen.Sagen Sie dann die Struktur von Einzelkettenproteinen voraus und führen Sie eine Proteinsprachenmodellierung durch.
Dann,Forscher haben AlphaMissense mithilfe menschlicher Proteine feinabgestimmt.Und das Ausgabeziel des Modells wurde festgelegt, nämlich die Pathogenität der Missense-Mutation.
Da sich unter den unbeobachteten Missense-Mutationen eine beträchtliche Anzahl gutartiger Mutationen befindet, diese jedoch beim Training als pathogene Mutationen klassifiziert werden, ist das AlphaMissense-Trainingsset sehr verrauscht.Um die Quantität und Qualität des Trainingssatzes zu verbessern, verwendeten die Forscher Selbstdestillation, um die Daten zu filtern.
Überprüfung klinischer Daten:Leistung in verschiedenen Datensätzen
Nach Abschluss der SchulungAlphaMissense wurde anhand kommentierter klinischer Daten (ClinVar-Datensatz), De-novo-Varianten bei Patienten mit seltenen Entwicklungsstörungen und MAVE-Ergebnissen in ProteinGym validiert.
Zunächst bewerteten die Forscher die Leistung von AlphaMissense anhand des ClinVar-Datensatzes. Nach der Analyse von 18.924 Mutationsstellen,Der auROC von AlphaMissense beträgt 0,940, was eine Verbesserung gegenüber dem vorherigen hochmodernen Evolutionsmodell (EVE) (0,911) darstellt.
Bei der klinischen Bewertung von Missense-Mutationen konzentriert man sich typischerweise auf Gene, die mit bestimmten Krankheiten in Zusammenhang stehen. Daher ist es besonders wichtig, zwischen gutartigen und pathogenen Missense-Mutationen in diesen Genen zu unterscheiden. Die Forscher verwendeten AlphaMissense, um 612 Gene in ClinVar zu analysieren.Sein auROC beträgt 0,950, was besser ist als die 0,921 von EVE.
Schließlich analysierten die Forscher die Vorhersageergebnisse von AlphaMissense im Deciphering Developmental Disorders (DDD)-Datensatz. Der auROC von AlphaMissense beträgt 0,809, was mit 0,797 von PrimateAI vergleichbar ist.
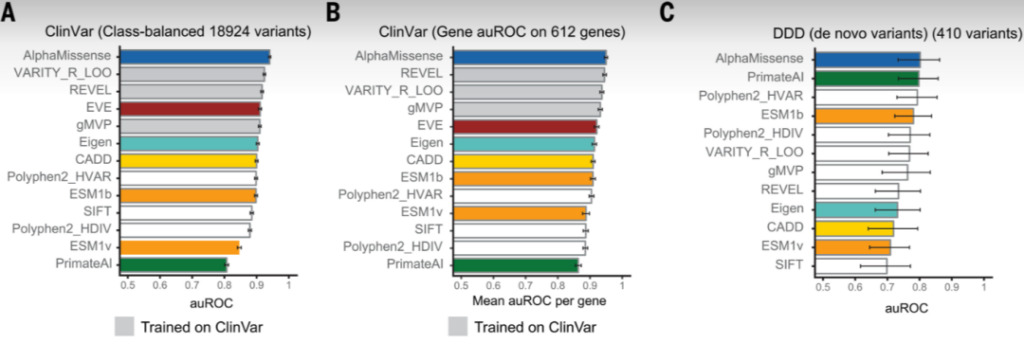
Abbildung 4: Leistungsvergleich von AlphaMissense und anderen Modellen in verschiedenen Datensätzen
A: Analyse von Mutationsstellen in ClinVar;
B: Analyse von Genen in ClinVar;
C: Analyse des DDD-Datensatzes.
Gleichzeitig sind die Vorhersageergebnisse von AlphaMissense für Krebs-Hotspots, ACMG (American College of Medical Genetics) und andere MAVE-Daten besser als die anderer Modelle.Die obigen Ergebnisse zeigen, dass AlphaMissense in mehreren Datensätzen bestehende Modelle übertrifft.
Gesamtvorhersageleistung:Spiegelt Proteinmutationstrends wider
Nach der Verifizierung von AlphaMissense anhand klinischer Daten,Die Forscher verwendeten AlphaMissense, um mögliche Mutationen von 216 Millionen Aminosäuren in 19.233 häufigen Proteinen beim Menschen vorherzusagen und erhielten schließlich Vorhersagen für 71 Millionen Missense-Mutationen.
Die Ergebnisse der Pathogenitätsvorhersage von AlphaMissense liegen zwischen 0 und 1. Je näher der Wert bei 1 liegt, desto höher ist die Wahrscheinlichkeit der Pathogenität. Da die überwiegende Mehrheit der Vorhersagen nahe bei 0 und 1 liegt, sind die Werte zwischen 0,2 und 0,8 wahrscheinlich weniger genau. Letztendlich teilten sie die Vorhersagen in drei Kategorien ein:Möglicherweise pathogen, möglicherweise gutartig und unbestimmt.
Um die Vorhersagekraft von AlphaMissense insgesamt zu bewerten, berechneten die Forscher die Pathogenität einzelner Aminosäuren für alle Proteine. Die Ergebnisse zeigen, dassMutationen in aromatischen Aminosäuren und Cystein führen eher zu Krankheiten, was mit den tatsächlichen Ergebnissen übereinstimmt.Denn diese beiden Aminosäuren spielen eine Rolle bei der Aufrechterhaltung der Proteinstruktur.
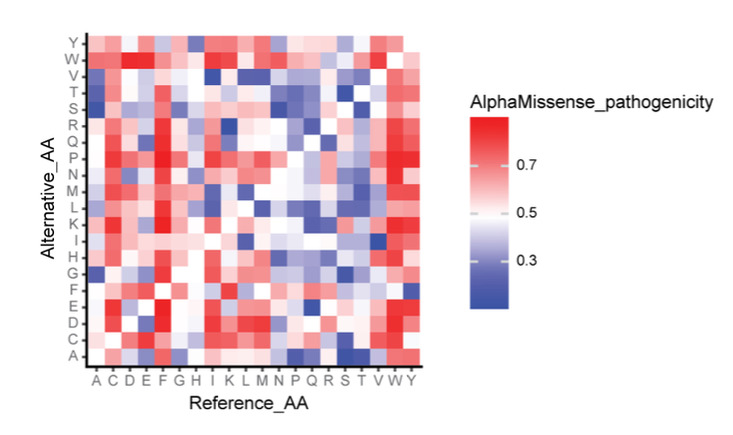
Abbildung 5: Heatmap der AlphaMissense-Vorhersageergebnisse,Die Farbblöcke stellen die durchschnittliche Pathogenität von 216 Millionen Aminosäureveränderungen im Proteom dar
Nachdem wir die Vorhersageergebnisse von AlphaMissense und die von AlphaFold vorhergesagten Proteinstrukturen visualisiert haben, können wir die Mutationstrends dieser Proteine erkennen.Beispielsweise entsprechen Regionen mit ungeordneter Proteinstruktur Regionen, in denen gutartige Mutationen auftreten, was mit den Vorhersageergebnissen der Proteomik übereinstimmt.
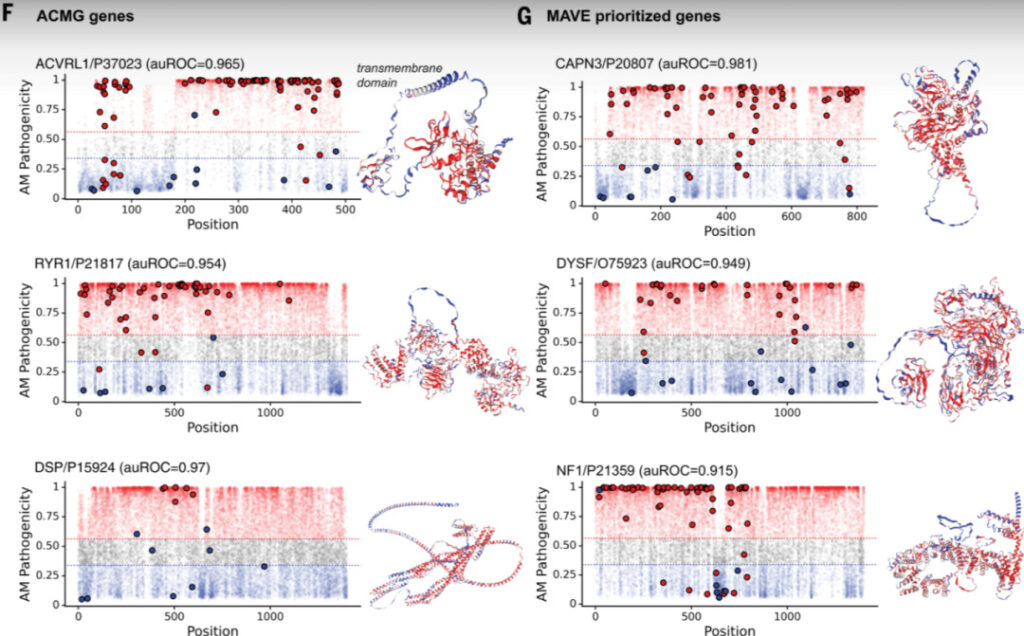
Abbildung 6: Visualisierungsergebnisse einiger Proteine in ACMG- und MAVE-Datensätzen
Links ist die von AlphaMissense vorhergesagte Pathogenität dargestellt. Missense-Mutationen, die Krankheiten verursachen können, sind rot, möglicherweise gutartige Missense-Mutationen blau und Mutationen, die in den ClinVar-Datensatz aufgenommen wurden, sind mit ausgefüllten Kreisen markiert. Rechts ist die von AlphaFold vorhergesagte Proteinstruktur. Verschiedene Farben zeigen die Pathogenität von Mutationen in dieser Region an, die AlphaMissense entspricht.
Vorhersagegenauigkeit:Konsistenz mit MAVE-Ergebnissen
Um die Konsistenz zwischen den Ergebnissen von AlphaMissense und MAVE zu untersuchen, analysierten die Forscher zwei Sätze von MAVE-Daten mithilfe von AlphaMissense.Im Vergleich zu anderen Vorhersagemethoden kommt AlphaMissense den MAVE-Daten am nächsten.
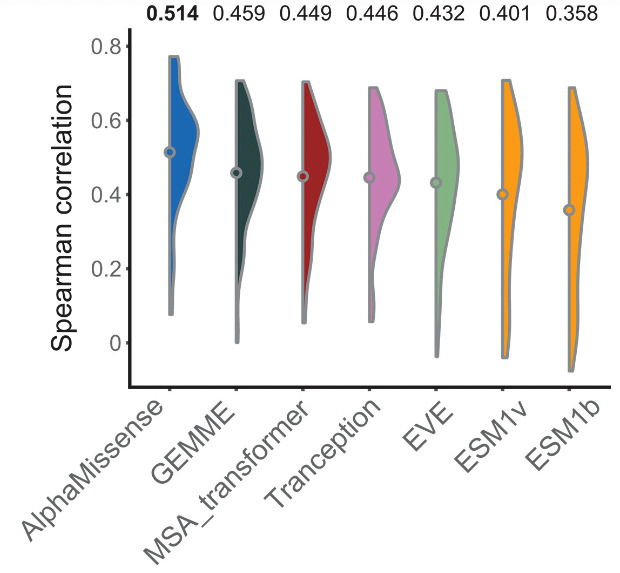
Abbildung 7: Spearman-Korrelationskoeffizient von AlphaMissense und anderen Modellen mit MAVE-Vorhersageergebnissen,Unter ihnen hat AlphaMissense das beste Ergebnis
Anschließend verglichen sie die Vorhersagedaten von AlphaMissense mit der durch Experimente bestätigten Pathogenität von Missense-Mutationen. Das SHOC2-Protein kann einen Komplex mit MRAS- und PP1C-Proteinen bilden, um den Ras-MAPK-Krebssignalweg zu aktivieren. AlphaMissense und MAVE haben den Zusammenhang zwischen dieser Mutation und Ras-Krebszellen vorhergesagt.Der erhaltene Spearman-Korrelationskoeffizient beträgt 0,47 und ist damit besser als bei anderen Modellen. (ESM1v: 0,41, ESM1b: 0,40, EVE: 0,32).
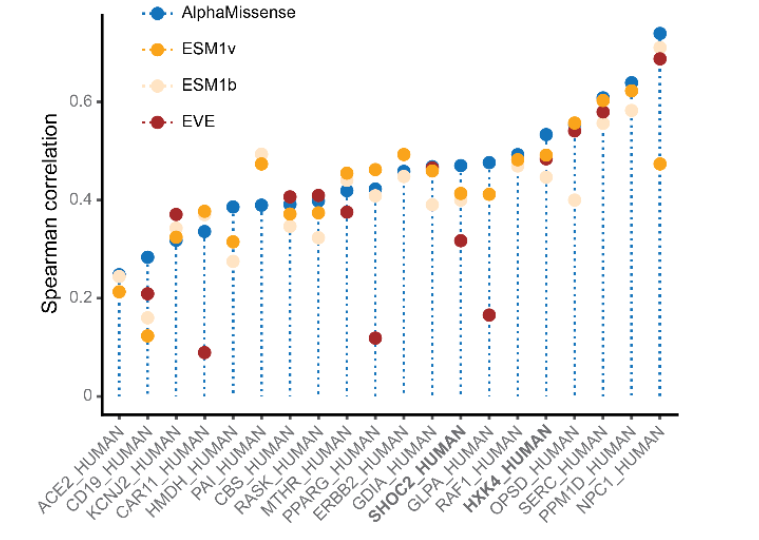
Abbildung 8: Vorhersageergebnisse verschiedener Modelle für Missense-Mutationen im MAVE-Datensatz
Darüber hinaus untersuchten die Forscher die Vorhersageergebnisse von AlphaMissense zur Pathogenität von Aminosäure-Missense-Mutationen in verschiedenen Regionen des SHOC2-Proteins. MAVE sagte voraus, dass Mutationen in den Aminosäuren 63–74 der ersten 80 Aminosäuren von SHOC2 pathogen sind, da diese Region über RVxF an das PP1C-Protein bindet. AlphaMissense ist das einzige Modell, das diese wichtige Region identifiziert.

Abbildung 9: AlphaMissense-Vorhersageergebnisse für das SHOC2-Protein
A: Vorhersageergebnisse verschiedener Modelle zur Pathogenität von Mutationen in den ersten 200 Aminosäuren des SHOC2-Proteins. Von oben nach unten sind dies die tatsächliche Situation (MAVE), AlphaMissense und EVE.
B: Das Strukturdiagramm des Komplexes, der aus dem SHOC2-Protein (rot und blau) und den Proteinen MRAS (gelb) und PP1C (gold) besteht.
Darüber hinaus kann AlphaMissense die Ergebnisse verschiedener Arten von Aminosäure-Missense-Mutationen widerspiegeln.Für das SHOC2-Protein kommen die Vorhersageergebnisse von AlphaMissense den tatsächlichen Ergebnissen am nächsten.
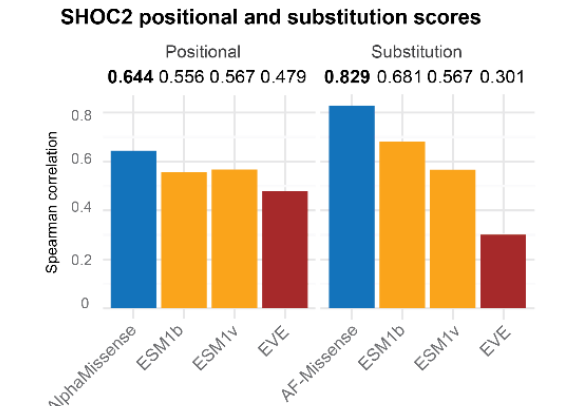
Abbildung 10: Korrelation zwischen verschiedenen Modellen zur Vorhersage der Pathogenität von Aminosäuremutationen in SHOC2- und MAVE-Ergebnissen
Zusammenfassend zeigen die obigen Ergebnisse, dass die Vorhersageergebnisse von AlphaMissense mit denen von MAVE vergleichbar sind und die Folgen von Missense-Genmutationen genau vorhersagen können.
Schließlich stellte Deepmind das Modell und die Vorhersageergebnisse der Community als Open Source zur Verfügung, in der Hoffnung, dass die Schlussfolgerungen der Forschung in anderen Disziplinen helfen könnten.
Modelllink:
https://github.com/deepmind/alphamissense
Genmutation: Unerreichbar und doch immer da
Wenn es um Genmutationen geht, denken wir sofort an gefährliche Elemente wie Röntgenstrahlen, radioaktive Strahlung, Nitrit oder Szenen aus den Filmen „Resident Evil“ und „Hulk“ und haben das Gefühl, dass diese zu weit weg von uns sind. Es stimmt, dass die Strahlung, der wir in unserem täglichen Leben ausgesetzt sind, sehr gering ist.Dennoch kommt es in jedem Moment unseres Lebens zu Genmutationen, die unser Leben tatsächlich verändern.
Im Leben sind wir unvermeidlich Strahlungsquellen ausgesetzt., wie Sonnenlicht. Die Strahlung im Sonnenlicht besteht aus ultravioletten Strahlen, die zu den krebserregenden Faktoren zählen. Daher erhöht eine langfristige Sonneneinstrahlung das Hautkrebsrisiko.
Auch ohne Exposition gegenüber StrahlungsquellenBei der Replikation, Transkription und Translation der DNA kommt es zwangsläufig zu Fehlern, die zu Genmutationen führen., aber diese Mutationen können gutartig sein oder mit der Zeit durch den Immunmechanismus beseitigt werden.
Gleichzeitig bringen Genmutationen aber auch Annehmlichkeiten in unser Leben.Besonders in der landwirtschaftlichen Produktion. Mutanten von Nutzpflanzen können den Ernteertrag steigern, die Salz- und Alkalitoleranz der Nutzpflanzen verbessern und sogar bei der Schädlingsbekämpfung helfen. Nach der Züchtung und Untersuchung dieser Mutanten können diese hervorragenden Eigenschaften erhalten bleiben und so die Nahrungsmittelproduktion gesteigert werden.

Abbildung 11: Verschiedene Sorten von Maismutanten
Allerdings gibt es zu viele Möglichkeiten für Genmutationen beim Menschen und unser derzeitiges Wissen ist nur ein Tropfen auf den heißen Stein. Mit AlphaMissense können wir relativ zuverlässige Vorhersagen über die Folgen von Genmutationen treffen und daraus Rückschlüsse ziehen.Vielleicht können wir die Mechanismen hinter genetischen und seltenen Krankheiten aufdecken und neue Methoden zur Krankheitsvorbeugung und -behandlung entwickeln.
Gleichzeitig stellt AlphaMissense auch Material für die Forschung in anderen Bereichen bereit. Vielleicht können wir bald beobachten, wie AlphaMissense genetische Mutationen bei anderen Arten interpretiert.Dann können wir Genmutationen sinnvoll nutzen und die Gentechnik unserem Leben mehr Vorteile bringen.
Referenzlinks:
[1]https://www.science.org/doi/10.1126/science.abj6987
[2]https://www.cshl.edu/discovery-of-new-stem-cell-pathway-indicates-route-to-much-higher-yields-in-maize-staple-crops/
Dieser Artikel wurde zuerst auf der öffentlichen HyperAI WeChat-Plattform veröffentlicht~








