Command Palette
Search for a command to run...
Strategie Zur Merkmalsauswahl: Neue Wege Zur Erkennung Von Brustkrebs-Biomarkern Finden

Inhalte im Überblick:microRNA (microRNA) ist eine Klasse kurzer, einzelsträngiger nicht-kodierender RNA-Transkripte. Diese Moleküle zeigen in einer Vielzahl bösartiger Tumoren ein unkontrolliertes Wachstum und wurden daher in vielen Studien der letzten Jahre als zuverlässige Biomarker für die Diagnose von Krebs identifiziert. Unter den verschiedenen pathologischen Analysen wird die Analyse der differentiellen Expression oft als wirksame Methode zum Erkennen wichtiger Biomarker angesehen. Forscher der Universität Neapel Federico II in Italien schlugen vor, dass eine auf maschinellem Lernen basierende Merkmalsauswahlstrategie für die Erkennung effektiver sein könnte, und empfahlen, die 20 von ihnen entdeckten microRNAs als diagnostische Biomarker für Brustkrebs zu verwenden.
Schlüsselwörter:Merkmalsauswahl microRNA Brustkrebs
Dieser Artikel wurde zuerst auf der öffentlichen HyperAI WeChat-Plattform veröffentlicht~
Laut der Ausgabe 2022 der „Leitlinien zur Behandlung von Brustkrebs“ der Nationalen GesundheitskommissionBrustkrebs ist einer der häufigsten bösartigen Tumoren bei Frauen und nimmt unter den bösartigen Tumoren bei Frauen die höchste Inzidenzrate ein.Laut Statistiken der Weltgesundheitsorganisation wurde im Jahr 2020 weltweit bei insgesamt 2,3 Millionen Frauen Brustkrebs diagnostiziert. Mit der kontinuierlichen Verbesserung der Behandlungsmethoden,Die Fünfjahresüberlebensrate bei Brustkrebs im Frühstadium kann 90% oder sogar mehr erreichen. Daher ist eine genaue Diagnose von Brustkrebs im Frühstadium besonders wichtig.
Veränderungen in der microRNA-Expression spielen nicht nur zahlreiche Schlüsselrollen in der Biologie, sie werden auch mit einer Reihe von Krebsarten in Zusammenhang gebracht, sodass sie als zuverlässiger mutmaßlicher diagnostischer Biomarker verwendet werden können. Forscher der Universität Neapel Federico II in Italien nutzten maschinelles Lernen, umDurch die Verwendung einer Merkmalsauswahlstrategie und die Analyse der Stabilität und Klassifizierungsleistung der drei MethodenEs wurde eine Reihe brustkrebsspezifischer diagnostischer Biomarker ermittelt und mutmaßliche Schlüsselgene für die Entwicklung und das Fortschreiten der Brustkrebserkrankung entdeckt.
Dieses Forschungsergebnis wurde kürzlich in den Proceedings der 18. Konferenz zu Methoden der Computational Intelligence in der Bioinformatik und Biostatistik (CIBB 2023) unter dem Titel „Robust Feature Selection strategy detects a panel of microRNAs as putative diagnostic biomarkers in Breast Cancer“ veröffentlicht.

Die Forschungsergebnisse wurden im CIBB 2023 veröffentlicht
Papieradresse:
https://www.researchgate.net/publication/372083934
Experimentübersicht
In dieser Studie fanden die Forscher heraus, dass mit Hilfe von drei Merkmalsauswahlmethoden (Gewinnrate, Random Forest und rekursive Merkmalseliminierung mittels Support Vector Machine) diagnostische molekulare Kombinationen effizienter extrahiert werden können. Sie enthüllten eine Gruppe von 20 Mikro-RNAs, darunter hsa-mir-337, hsa-mir-378c und hsa-mir-483, die unter den aktuellen diagnostischen Biomarkern für Brustkrebs in der medizinischen Gemeinschaft keine große Beachtung gefunden haben. Mit dieser Methode können gesunde Proben von Tumorproben unterschieden werden. Im Vergleich zur häufig verwendeten Methode des differentiellen Ausdrucks ist die Klassifizierungsleistung besser und es ist einfacher, Merkmale zu identifizieren, die leicht unterschätzt oder sogar ignoriert werden.
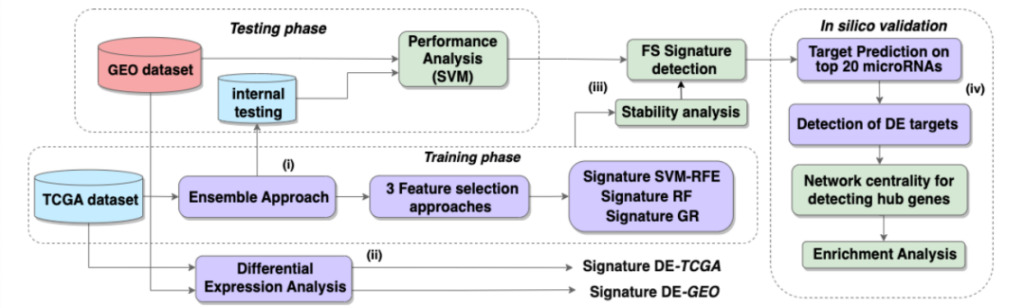
Abbildung 1: Pipeline-Übersicht
Der Arbeitsablauf besteht aus vier Hauptschritten:
(ich) Ensemble-FS-Berechnungen auf der Trainings-TCGA-Teilmenge
(ii) Differenzielle Expressionsanalyse von TCGA/GEO-Datensätzen
(iii) Vergleichen Sie die Klassifizierungsleistung der Analyse differentieller Ausdrücke und die Ergebnisse der Merkmalsauswahl und bewerten Sie die Stabilität der Methoden zur Merkmalsauswahl
(iv) Mithilfe einer Computersimulation wurden die 20 wichtigsten Mikro-RNAs der ausgewählten Signatur verifiziert und Hub-Genziele erkannt.
Details des Experiments
Datensatz
Die experimentellen Datenquellen umfassen zwei Kanäle:Das TCGA-BRCA-Projekt auf der offiziellen Website des US GDC und des Gene Expression Omnibus (GEO)-Datenrepository (GSE97811).
Das Experimentalteam sammelte insgesamt 1.881 microRNA-Seq-Daten aus dem GDC TCGA-BRCA-Projekt und teilte sie im Verhältnis 8:2 in Trainings- und Testsätze auf. Die Daten wurden mit 300 festen Primärtumorproben (T) und 101 normalen angrenzenden Tumorproben (NAT) in Verbindung gebracht, sowohl aus duktalem als auch aus lobulärem Brustgewebe. Bevor Sie die Merkmalsauswahl anwenden,Diese Daten wurden einer varianzstabilisierenden Normalisierung unterzogen.
Gleichzeitig luden die Forscher einen Microarray-Datensatz mit 2.565 microRNAs aus der GEO-Datenbank (GSE97811) herunter.Es wird als Validierungssatz für dieses Experiment verwendet.Der Datensatz umfasst 16 normale Proben und 45 Tumorproben, anschließend wird eine Datenimputation durchgeführt.
Da die GEO-Daten (der Validierungssatz in diesem Experiment) die Expression reifer microRNAs beinhalten und die TCGA-Daten (der Trainingssatz und Testsatz in diesem Experiment) die Vorläuferform enthalten, wählten die Forscher zur Vereinheitlichung der Daten nur alternative reife microRNAs mit höheren durchschnittlichen Zählwerten als ihre Gegenstränge in den GEO-Datenproben aus. gleichzeitig wurden die Namen der microRNAs in die entsprechenden Namen der Vorläuferformen umgewandelt.
Nach diesem VorgangDie Dimensionalität der GEO-Daten (Validierungssatz) wurde auf 1.361 MikroRNAs reduziert und die entsprechenden TCGA-RNA-Seq-Daten wurden ebenfalls gesammelt, darunter insgesamt 20.404 Gene.
1. Merkmalsauswahlmethode und Ensemble-Programmanwendung
Die Forscher wählten drei Methoden zur Merkmalsauswahl für den Vergleich mit der Analyse der differentiellen Expression aus.Sie sind Verstärkungsverhältnis, Random Forest und SVM-RFE (Support Vector Machine Recursive Feature Elimination).Drei Methoden wurden auf 500 Teilmengen von TCGA-Expressionsdaten der microRNA-Seq angewendet, um ein robustes Merkmalspanel zu identifizieren, mit dem zwischen normalen und Tumorproben unterschieden werden kann. In den beobachteten Ergebnissen wurden die Daten im Verhältnis 8:2 in Trainingsdaten und Testdaten aufgeteilt. Anschließend wurden die Daten erneut abgetastet und gebootet, um sie dem Data Perturbation Ensemble-Verfahren anzupassen. Jede Berechnung gibt 500 microRNA-Vektoren zurück, die in absteigender Reihenfolge nach „Wichtigkeitsbewertung“ sortiert sind.
|Bemerkungen:Der Wichtigkeitswert stellt den Einfluss jedes Merkmals in der vom Algorithmus berechneten Klassifizierung dar.
Je höher der Wichtigkeitswert, desto niedriger ist der dem Feature zugewiesene Rang.Anschließend verwendeten die Forscher ein Aggregationsverfahren, um für jede Merkmalsauswahlmethode eine Konsenssignatur abzuleiten, und behielten schließlich die 200 wichtigsten Merkmale für jede Gruppe von Mikro-RNAs bei.
2. Stabilitätstest
Der Kuncheva-Index (KI) und der Prozentsatz überlappender Gene/Merkmale (POG) wurden verwendet, um die Konsistenz der Merkmalsauswahlmethoden zu bewerten, und die Stot-Statistik (paarweise Messung des KI) wurde verwendet, um die Stabilität aller Methoden zu bestimmen.Diese Statistiken werden mit zunehmender Signaturlänge berechnet.Die Anzahl der Features beginnt bei 2 und endet bei 200, und jede Neuberechnung erhöht sich um 2 Einheiten.

Stot-Statistikformel
3. Differentialexpressionsanalyse und DE-Signatur
Die Analyse der differentiellen Expression wurde an TCGA-Datensätzen (einschließlich microRNA-Seq und RNA-Seq) durchgeführt, beginnend mit den Rohzählungen, unter Verwendung des Exact-Tests und anschließender Beibehaltung der DE-Funktionen mit FDR <= 0,01 und einem Log2FC-Schwellenwert von |0,5|.Um die Signatur der DE-microRNA zu erhalten, wurden die Log2FC-Werte in absolute Werte umgewandelt und die microRNAs (die ersten 200 Merkmale wurden beibehalten) in absteigender Reihenfolge der Abs (Log2FC) sortiert.
Der GEO-Validierungssatz wurde mit Limma differenziell ausgedrückt, und die Parameter und Verfahren zum Erhalten von DE-Signaturen in diesem Datensatz stimmten mit denen des TCGA-Datensatzes überein.
4. Klassifizierungsleistungsanalyse
Um die Fähigkeit jeder Signatur zu bestimmen, gesunde Menschen von Krebspatienten zu unterscheiden,Die Forscher führten eine prädiktive Analyse von vier Signaturen (einschließlich Merkmalsauswahlfeldern und Feldern für differenzielle Expression) sowohl im Test-Subset (TCGA) als auch im Validierungsset (GEO) durch.
Schließlich werden die durchschnittliche Genauigkeit (ACC), die K-Statistik (KK) und der Matthews-Korrelationskoeffizient (MCC) für jede Falte und mehrere Längen jeder Signatur berechnet.
5.SVM-RFE microRNA-Signatur-Zielerkennung
Um potentielle Genziele von microRNA zu identifizieren,Die Forscher führten die folgenden Operationen durch:
1. Die 20 wichtigsten SVM-RFE-Mikro-RNAs wurden danach klassifiziert, ob sie in Tumorproben hoch- oder herunterreguliert waren.
2. Anhand der RNA-Seq-Daten wurde eine Analyse der differentiellen Expression durchgeführt, um differentiell exprimierte Gene zu erkennen (FDR <= 0,05).
3. Um die Expression von microRNAs mit unterschiedlich exprimierten Genen zu vergleichen, wurde eine Spearman-Korrelationsanalyse durchgeführt. Dabei wurden nur Up-Gene, die negativ mit Down-microRNAs korreliert waren, und Down-Gene, die negativ mit Up-microRNAs korreliert waren, beibehalten (rho <= -0,5).
4. Alle validierten microRNA-Genziele wurden gesammelt und nur diejenigen beibehalten, die auch eine DE-Korrelation zeigten.
6. Netzwerkzentralität und Identifizierung von Hub-Genen
Korrelationsmatrix (Spearman) ausgewählter dysregulierter Gene,Und verwenden Sie es, um ein Gennetzwerk mit Graphstruktur aufzubauen:Hub-Gene mit Kleinbergs Hub-Zentralitäts-Score > 75, rho > 0,8 oder rho < -0,6 wurden beibehalten. An den Hub-Genen wurde eine Genanreicherungsanalyse (ORA) durchgeführt, um die am stärksten angereicherten Pfade aus der REACTOME-Datenbank zu untersuchen. Der FDR-angepasste p-Wert-Schwellenwert wurde auf 0,005 festgelegt.
Experimentelle Ergebnisse
Das Experiment zeigte, dass nach Anwendung der drei Merkmalsauswahlmethoden 500 microRNA-Signaturen in absteigender Reihenfolge der Wichtigkeitsbewertung zurückgegeben wurden und nach der Aggregation drei Konsenspanels erhalten wurden. Bemerkenswerterweise erschienen die drei wichtigsten microRNAs (hsa-mir-139, hsa-mir-96 und hsa-mir-145) in allen Panels, was die Bedeutung dieser Moleküle bei der Unterscheidung von Tumorproben von gesunden Proben verdeutlicht.
Schlussfolgerung 1: SVM-RFE hat die höchste Stabilität
Aus der Berechnung von KI und POG im Konsensusgremium,Die SVM-RFE-Methode ist die stabilste und am stärksten ausgeprägt, wenn die Signaturlänge 20 Merkmale erreicht. Ebenso zeigen die Ergebnisse des Stot-Index, dass die SVE-RFE-Methode die höchste Stabilität aufweist.
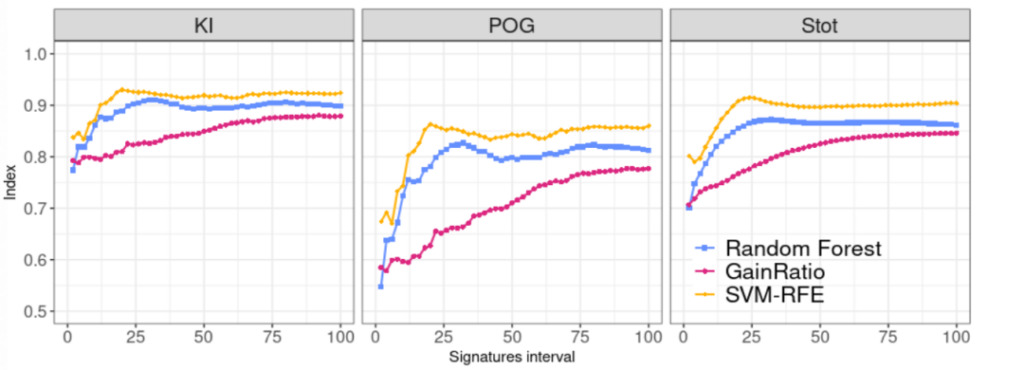
Abbildung 2: Vergleich des Stabilitätsindex dreier Methoden zur Merkmalsauswahl
Blau:Zufälliger Wald
Rosa:Verstärkungsverhältnis
Gelb:SVM-RFE (Rekursive Merkmalseliminierung bei Support Vector Machines)
|Schlussfolgerung 2: Die SVM-RFE-Signatur ist bei der Klassifizierung leistungsfähiger als die Signatur des differentiellen Ausdrucks
Nach der Analyse der Klassifizierungsleistung aller einzelnen Panels zeigten sowohl das Testset (TCGA) als auch das Validierungsset (GEO), dass die durch SVM-RFE erhaltene Signatur die höchste Vorhersagekraft hatte.
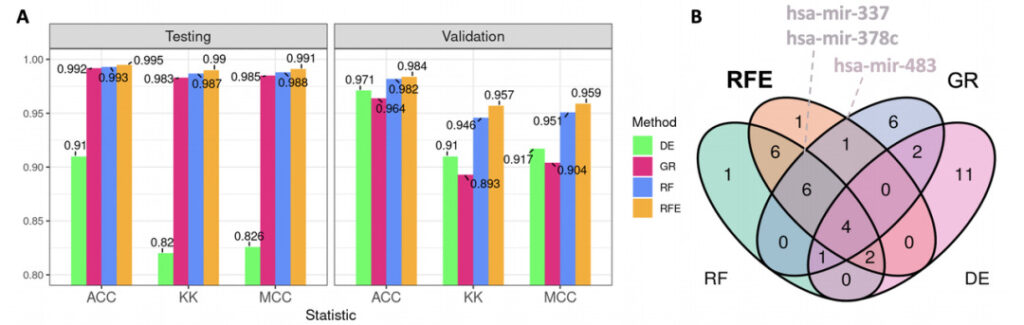
Abbildung 3: Klassifizierungsleistung der Top 20 microRNAs und Venn-Diagramm
A:Das Balkendiagramm zeigt die durchschnittliche Statistik, die auf der Grundlage der Testteilmenge und des externen Validierungs-GEO-Datensatzes berechnet wurde.
ACC:Genauigkeit
KK:K-Statistik
Kundencenter:Matthews-Korrelationskoeffizient
Grün:DE (Differentialexpressionsanalyse, die Kontrollmethode in diesem Experiment)
Rosa:GR (Verstärkungsverhältnis)
Blau:RF (Random Forest)
orange Farbe:RFE (SVM-RFE, Support Vector Machine Recursive Feature Elimination)
B:Abbildung 4: Venn-Diagramm der 20 wichtigsten microRNAs für jede Signatur, mit einigen interessanten microRNAs aus den Top 20 des SVM-RFE-Panels mit der Bezeichnung – hsa-mir-337, hsa-mir-378c und hsa-mir-483. Diese drei Mikro-RNAs tauchten in allen drei Merkmalsauswahlmethoden auf, ihre Zuverlässigkeit als diagnostischer Beweis konnte in aktuellen Studien zu Brustkrebs jedoch noch nicht vollständig geklärt werden.
|Schlussfolgerung 3: Netzwerkanalyse enthüllt potenzielle Schlüsselgene in der Krankheitsentwicklung
Experimente haben gezeigt, dass CDC25, TPX2 und KIF18B in Stammzellen verschiedener Krebsarten und bei Patienten mit dreifach negativem Brustkrebs stark exprimiert werden und dass eine Herunterregulierung von TGFBR2 mit dem Fortschreiten der Krebserkrankung in Zusammenhang steht.
MicroRNA: Ein weiterer idealer Kandidat für die Früherkennung von Brustkrebs
Herkömmliche Brustkrebs-Screening-Methoden basieren immer noch auf Röntgenaufnahmen und Gewebebiopsien, die kein tieferes und umfassenderes Verständnis des gesamten Krebsgenoms ermöglichen. Diese Methode ist nicht nur hochgradig invasiv, teuer und anfällig für Nebenwirkungen, sondern führt auch häufig zu falsch positiven oder falsch negativen Ergebnissen. Es ist schwierig, die Genauigkeit der Brustkrebsfrüherkennung und das Patientenerlebnis zu verbessern.Es besteht weiterhin Bedarf an der Entwicklung neuer Strategien zur Bekämpfung der Belastung durch Brustkrebs.
Seit seiner Entdeckung im Jahr 1993 hat micorRNA unser Verständnis von Krebs kontinuierlich vertieft und großes Potenzial als zuverlässiger Biomarker für die Brustkrebsdiagnose gezeigt.
MicroRNA ist eine kleine nicht-kodierende RNA mit einer Länge von etwa 19–25 nt, die eine Vielzahl von Zielgenen regulieren kann.Beteiligt an der Regulierung einer Vielzahl biologischer und pathologischer Prozesse.Unter Berücksichtigung der Entstehung und Entwicklung von Krebs soll es die Einschränkungen der derzeitigen Röntgenbildgebung und Gewebebiopsie als gängige Diagnosemethoden für das Brustkrebs-Screening in der klinischen Praxis ausgleichen.
Allerdings sind ausgereifte klinische Anwendungen von microRNA noch nicht vollständig entwickelt und es wurde noch kein Sicherheitsbewertungssystem für die Verwendung von microRNA etabliert.Es wird wahrscheinlich einige Zeit dauern, bis microRNA zur gängigen diagnostischen Grundlage für Krebs wird.
Referenzartikel:
[1]https://www.who.int/zh/news-room/fact-sheets/detail/breast-cancer
[2]https://guide.medlive.cn/guideline/25596
[3]https://www.abcam.cn/kits/micrornas-as-biomarkers-in-cancer-1
[4]https://caivd-org.cn/webfile/file/20220508/20220508153691029102.pdf
[5]https://www.sohu.com/a/318088245_100120288
Dieser Artikel wurde zuerst auf der öffentlichen HyperAI WeChat-Plattform veröffentlicht~








