HyperAI
Command Palette
Search for a command to run...
LAMMPS:以单晶铝为例,模拟材料单轴拉伸
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)是一种经典的分子动力学仿真代码,专注于材料建模。它旨在在并行计算机上高效运行,并且易于扩展和修改。 LAMMPS 最初由美国能源部机构桑迪亚国家实验室开发,现在包括来自许多机构的许多研究小组和个人的贡献。 LAMMPS 的大部分资金来自美国能源部(DOE)。 LAMMPS 是根据 GNU 公共许可证版本 2(GPLv2)的条款分发的开源软件。
一、教程内容
在本次教程中,我们通过改变材料的晶格常数,实现模拟对施加材料单轴应变的情况,后续再计算并绘制材料的应变应力曲线。通过本教程的学习您将学会:
- 使用 npt 让系统结构达到弛豫
- 使用 fix 结合 variable 命令改变晶格常数
- 使用 compute 命令计算系统应力

二、输入文件
- Al99.eam.alloy 材料的 eam 势
- in.txt 输入文件
- pl.py 绘制图像脚本
三、输入文件解析
系统初始化与基础设置
units metal # 采用金属单位制(长度:Å,时间:ps,质量:g/mol,能量:eV 等)
dimension 3 # 三维空间模拟
boundary p p p # 三个方向均为周期性边界条件(模拟无限大晶体)
atom_style atomic # 原子类型为「基本原子」,仅包含位置和类型信息
variable latparam equal 4.05 # 定义铝的晶格常数为 4.05Å(FCC 铝的典型值)
原子结构构建
lattice fcc ${latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1 # 定义 FCC 晶格 # 晶格常数为 ${latparam},晶向沿 x[100] 、 y[010] 、 z[001](标准笛卡尔坐标系)region whole block 0 10 0 10 0 10 # 定义模拟区域:一个立方体,在 x 、 y 、 z 方向各包含 10 个晶格常数长度
create_box 1 whole # 创建容纳原子的「盒子」,仅包含 1 种原子类型,边界为上述 region 定义的范围
create_atoms 1 region whole # 在盒子内填充类型为 1 的原子,按 FCC 晶格排列
原子间相互作用势设置
pair_style eam/alloy # 采用嵌入原子法(EAM)合金势描述原子间相互作用(适合金属体系)
pair_coeff * * Al99.eam.alloy Al # 配置势函数参数:
# - 第一个*:所有原子类型(此处仅 1 种)
# - 第二个*:所有原子类型的相互作用
# - Al99.eam.alloy:势函数文件(预训练的铝势)
# - Al:指定原子类型 1 对应元素为铝
计算量定义(用于后续分析)
compute csym all centro/atom fcc # 计算每个原子的中心对称参数(用于识别 FCC 结构完整性,缺陷处会偏离)
compute peratom all pe/atom # 计算每个原子的势能(用于分析能量分布)
平衡模拟阶段(消除初始应力,达到稳定状态)
reset_timestep 0 # 重置时间步计数器为 0(平衡阶段作为新起点) timestep 0.001 # 时间步长为 0.001ps(即 1fs,金属模拟常用精度) #初始化原子速度:温度 300K(室温),随机种子 12345,消除整体动量(mom yes)和旋转(rot no)
velocity all create 300 12345 mom yes rot no #施加 NPT 系综(恒温、恒压、恒粒子数)进行平衡:
#- temp 300 300 1:温度维持 300K,热浴阻尼系数 1(单位:ps)
#- iso 0 0 1:各向同性压力控制,目标压力 0bar,压力阻尼系数 1(单位:ps)
#- drag 1:原子运动阻尼系数(用于稳定系综)
fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1 #热力学输出设置:
thermo 1000 #每 1000 步输出一次热力学信息
thermo_style custom step lx ly lz press pxx pyy pzz pe temp #输出内容:
#步数、盒子尺寸(x/y/z)、总压力、压力张量分量、势能、温度 run 20000 #运行 20000 步平衡模拟(总时间 20000×0.001ps=20ps)
unfix 1 #平衡结束,移除 NPT 约束#记录平衡后的 x 方向盒子长度(用于后续计算应变)
variable tmp equal "lx" #临时变量存储当前 x 方向长度
variable L0 equal ${tmp} #定义 L0 为初始长度(拉伸前的平衡长度)
print "Initial Length, L0: ${L0}" #输出初始长度,便于验证
单轴拉伸变形阶段
reset_timestep 0 #重置时间步计数器为 0(拉伸阶段作为新起点) #重新施加 NPT 系综,但仅控制温度和横向压力:
#- temp 300 300 1:维持温度 300K
#- y 0 0 1 z 0 0 1:y 和 z 方向压力保持 0bar(横向自由,模拟单轴拉伸)
fix 1 all npt temp 300 300 1 y 0 0 1 z 0 0 1 drag 1 #定义拉伸速率:
variable srate equal 1.0e10 #应变速率为 1e10 /s(分子动力学常用高应变速率,远高于实验)
variable srate1 equal "v_srate / 1.0e12" #转换为 LAMMPS 单位(应变速率单位:ps⁻¹,1e10/s = 1e-2 ps⁻¹)#施加 x 方向拉伸变形:
#- deform 1 x:沿 x 方向变形,变形组为 1(所有原子)
#- erate ${srate1}:按上述应变速率拉伸
#- units box:变形基于盒子尺寸
#- remap x:原子坐标随盒子拉伸同步更新(避免原子「跑出」盒子)
fix 2 all deform 1 x erate ${srate1} units box remap x
拉伸过程数据输出
#定义应变和应力变量: variable strain equal "(lx - v_L0)/v_L0" #工程应变 =(当前 x 长度 - 初始长度)/ 初始长度 variable p1 equal "v_strain" #p1 对应工程应变(无量纲) variable p2 equal "-pxx/10000" #x 方向应力(GPa):pxx 为 LAMMPS 内置压力张量(单位 bar),负号转为拉应力,除以 10000 转换为 GPa variable p3 equal "-pyy/10000" #y 方向应力(GPa) variable p4 equal "-pzz/10000" #z 方向应力(GPa) #输出应变-应力数据到文件:
#- 每 100 步输出一次 p1(应变)、 p2(x 应力)、 p3(y 应力)、 p4(z 应力)
#- 保存到 Al_tens_100.def1.txt,不在屏幕显示
fix def1 all print 100 "${p1} ${p2} ${p3} ${p4}" file Al_tens_100.def1.txt screen no #输出原子轨迹文件(用于可视化):
#- 每 100 步保存一次,文件名为 md.lammpstrj
#- 包含原子 ID 、类型、 x/y/z 坐标
dump trace all custom 100 md.lammpstrj id type x y z #热力学信息输出设置:
thermo 1000 #每 1000 步输出一次
thermo_style custom step v_strain temp v_p2 v_p3 v_p4 ke pe press #输出步数、应变、温度、三方向应力、动能、势能、总压力run 30000 #拉伸阶段运行 30000 步(总时间 30ps,累积应变约 0.3)
模拟结束
print "All done" # 输出完成信息,提示模拟结束
四、上手模拟
1. 克隆本容器后进入容器设置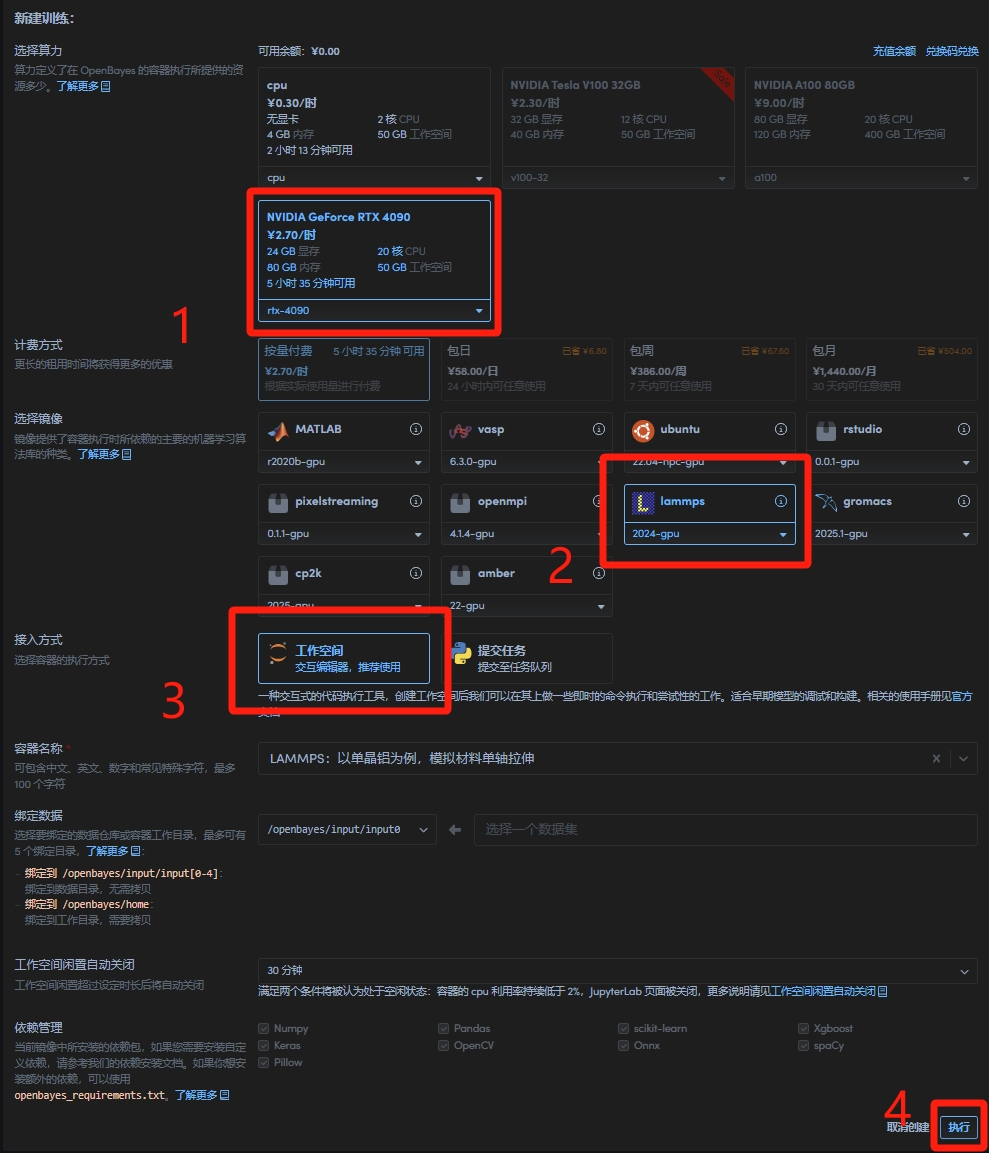
2. 待加载完毕后打开工作空间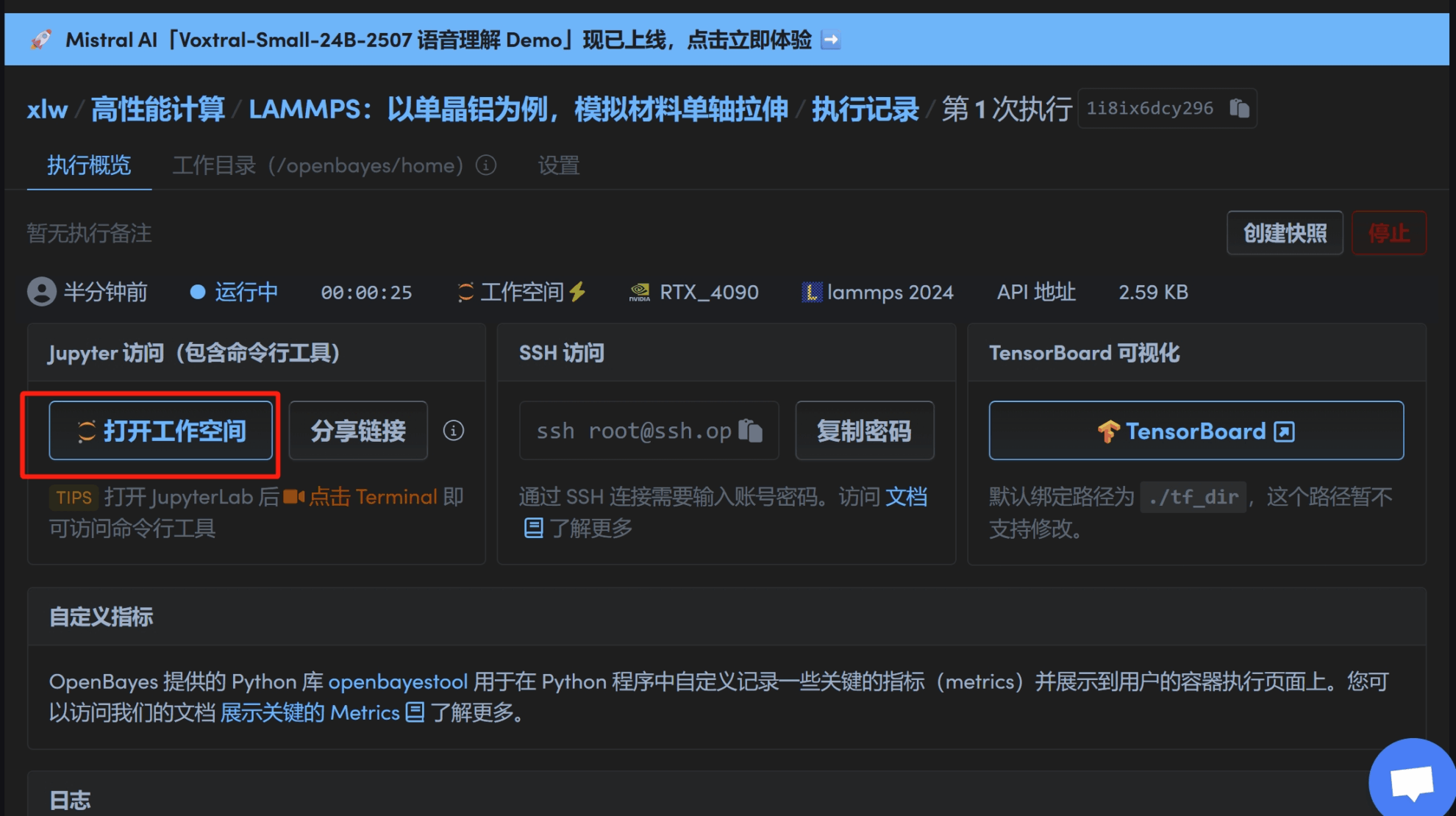
3. 随后打开终端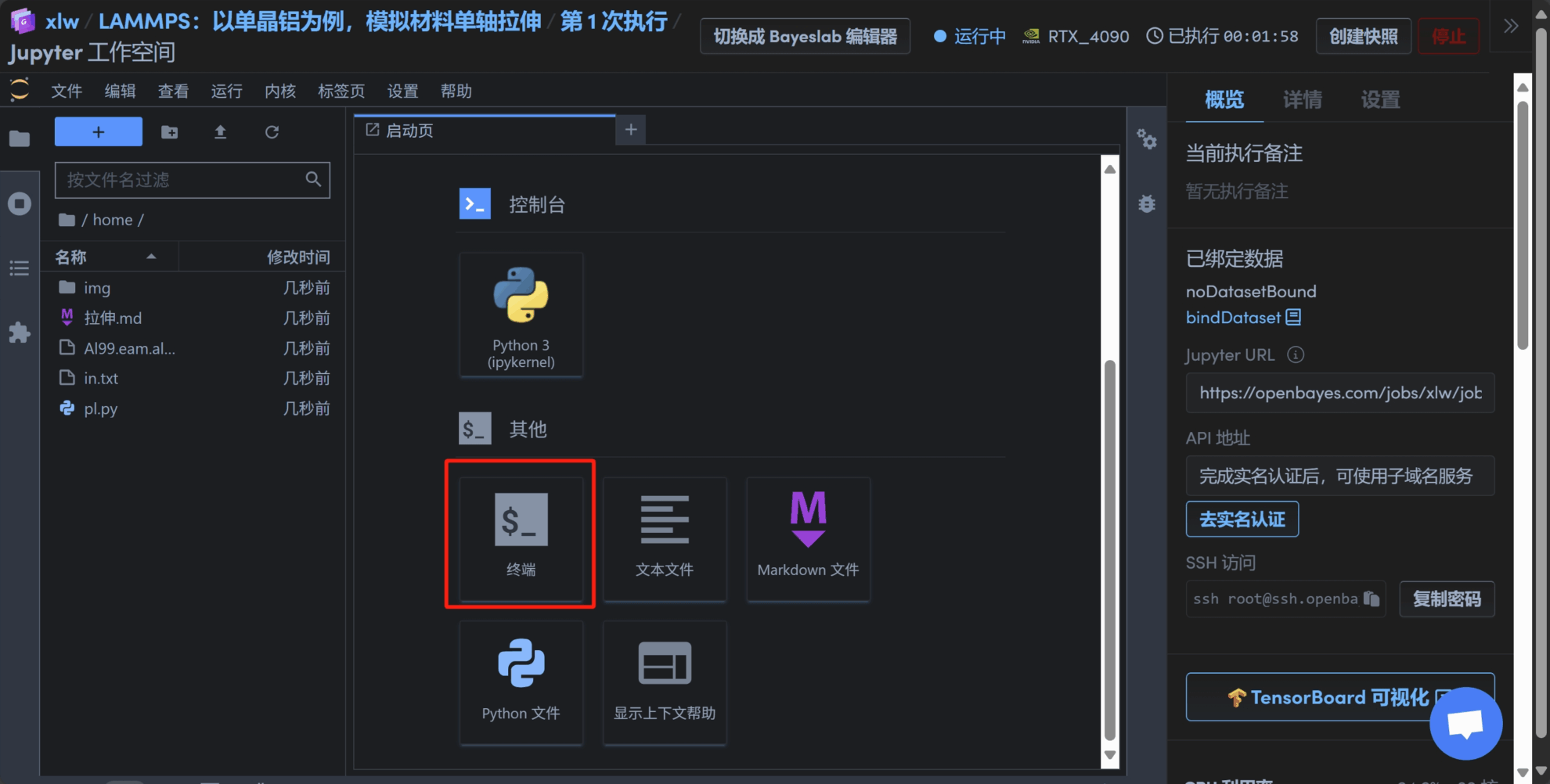
4. 输入以下命令运行 lammpsmpirun --allow-run-as-root -np 2 lmp -sf gpu -pk gpu 1 < in.txt 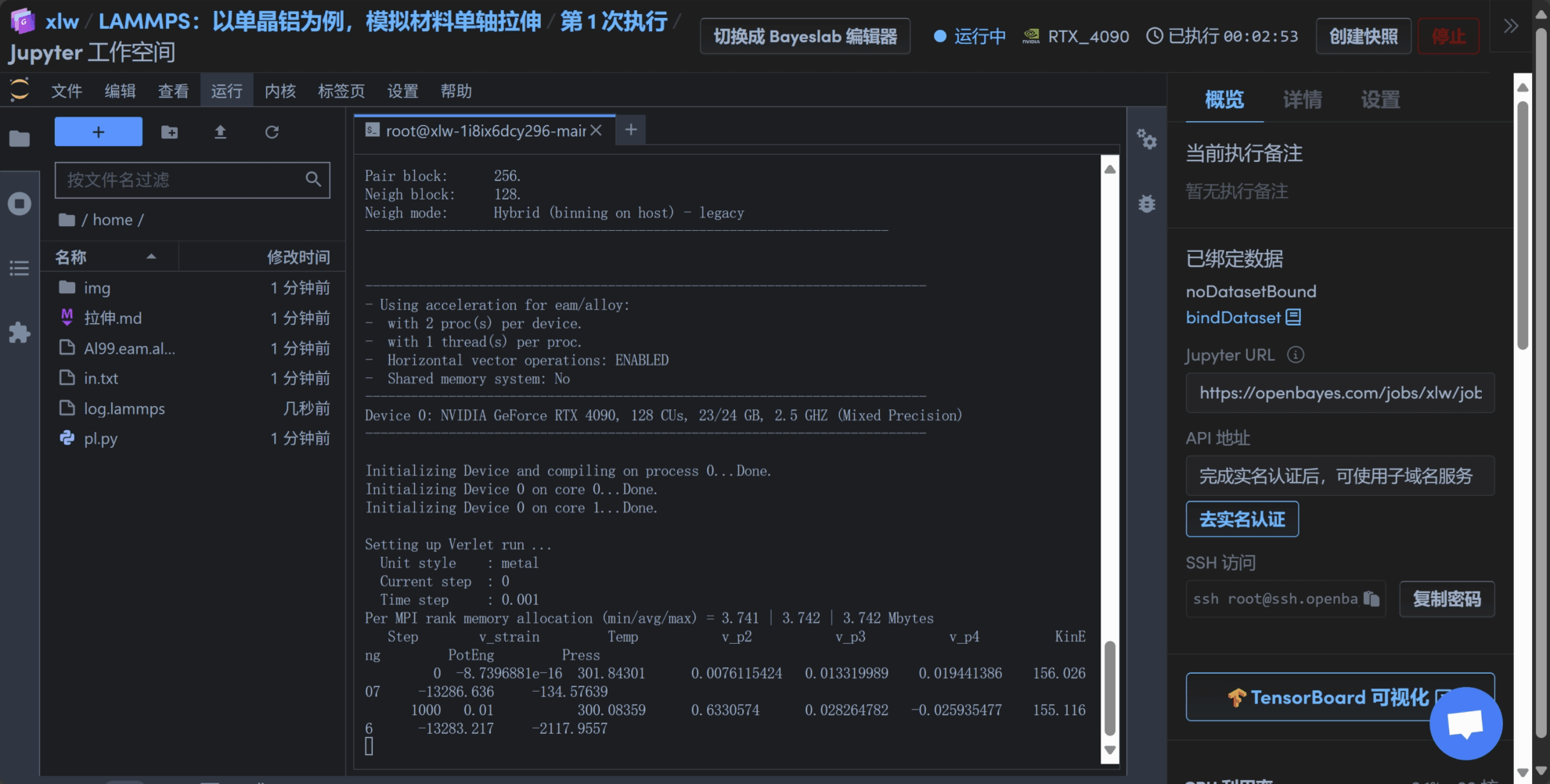
五、文件输出
- Al_tens_100.def1.txt:应变 – 应力曲线数据,可用于分析弹性模量、屈服强度等力学性能。
- md.lammpstrj:原子轨迹文件,可通过 VMD 、 Ovito 等软件可视化拉伸过程中的晶体结构演变
六、绘制应变-应力图像
1. 在终端输入以下命令python pl.py2. 打开绘制的图片
3. 可以得到曲线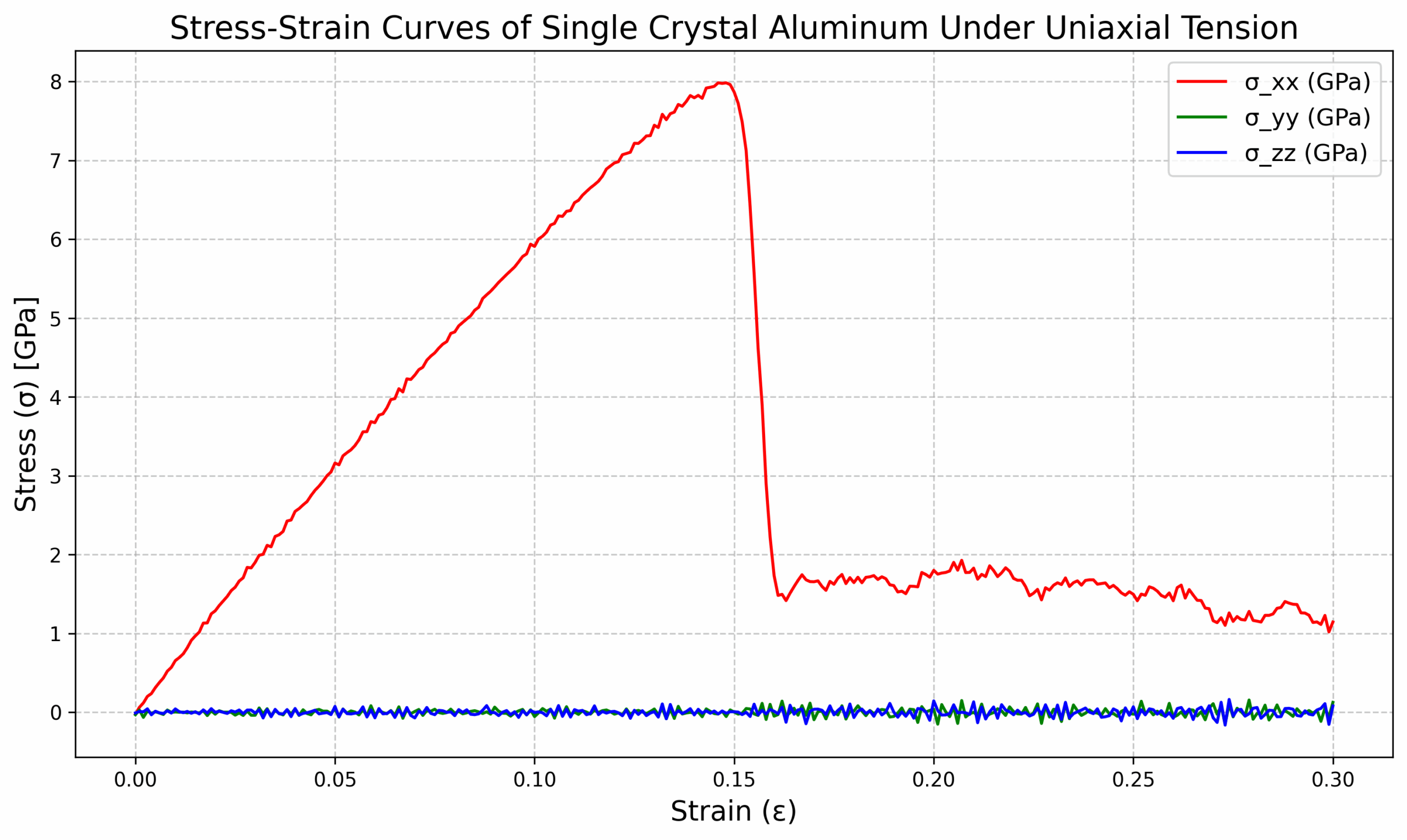
该教程由社区用户贡献,仅供交流学习使用。如内容涉及侵权,请联系邮箱 [email protected] 以便及时审查和下架。