Command Palette
Search for a command to run...
单细胞转录组测序单样本教程:质控、聚类、(差异)基因展示
单细胞转录组测序分析教程
本次示例数据采用发表于 2024 年 7 月 Nature medicine 的论文 Personalized neoantigen vaccine and pembrolizumab in advanced hepatocellular carcinoma: a phase 1/2 trial 中,6 号患者的人外周血单核细胞的单细胞测序数据 (Yarchoan M et al. Nat Med. 2024;30:1044–1053.) 。
单细胞测序简介
单细胞测序是一项精密的生物技术,它允许科学家在单个细胞层面上深入分析基因表达。这个过程通常从组织样本开始,首先将其解离成单个细胞,形成单细胞悬液。接着,利用油包水技术,将每个细胞单独封装,确保它们在后续的分析中保持独立。
在单细胞测序中,每个细胞的 mRNA 分子都会被标记上一个独特的分子标识符,这个过程称为 UMI(Unique Molecular Identifier) 标记。同时,每个细胞也会被赋予一个相同的 barcode 标记,这个 barcode 用于在数据分析阶段追踪每个细胞的来源。通过这种方式,研究人员能够精确地测量每个细胞中的 mRNA 信息,从而了解每个细胞的基因表达状况。
1.barcode 作用:记录细胞信息
2.UMI 在单细胞测序中的应用:通过为每个 mRNA 分子提供唯一标识,不仅提高了基因表达量测量的准确性,而且减少了 PCR 扩增过程中可能出现的误差,从而增强了整个测序过程的可靠性和有效性。
基本流程
单细胞 RNA 测序(scRNA-seq)的流程主要包括单细胞分离与捕获、细胞裂解、逆转录(将 RNA 转化为 cDNA)、 cDNA 扩增和文库制备
图源:Kulkarni, A., et al. (2019). “Beyond bulk: a review of single cell transcriptomics methodologies and applications” Curr Opin Biotechnol 58: 129-136
运行步骤
下面正式进入教程实操部分。
启动容器并上传数据
1. 启动容器
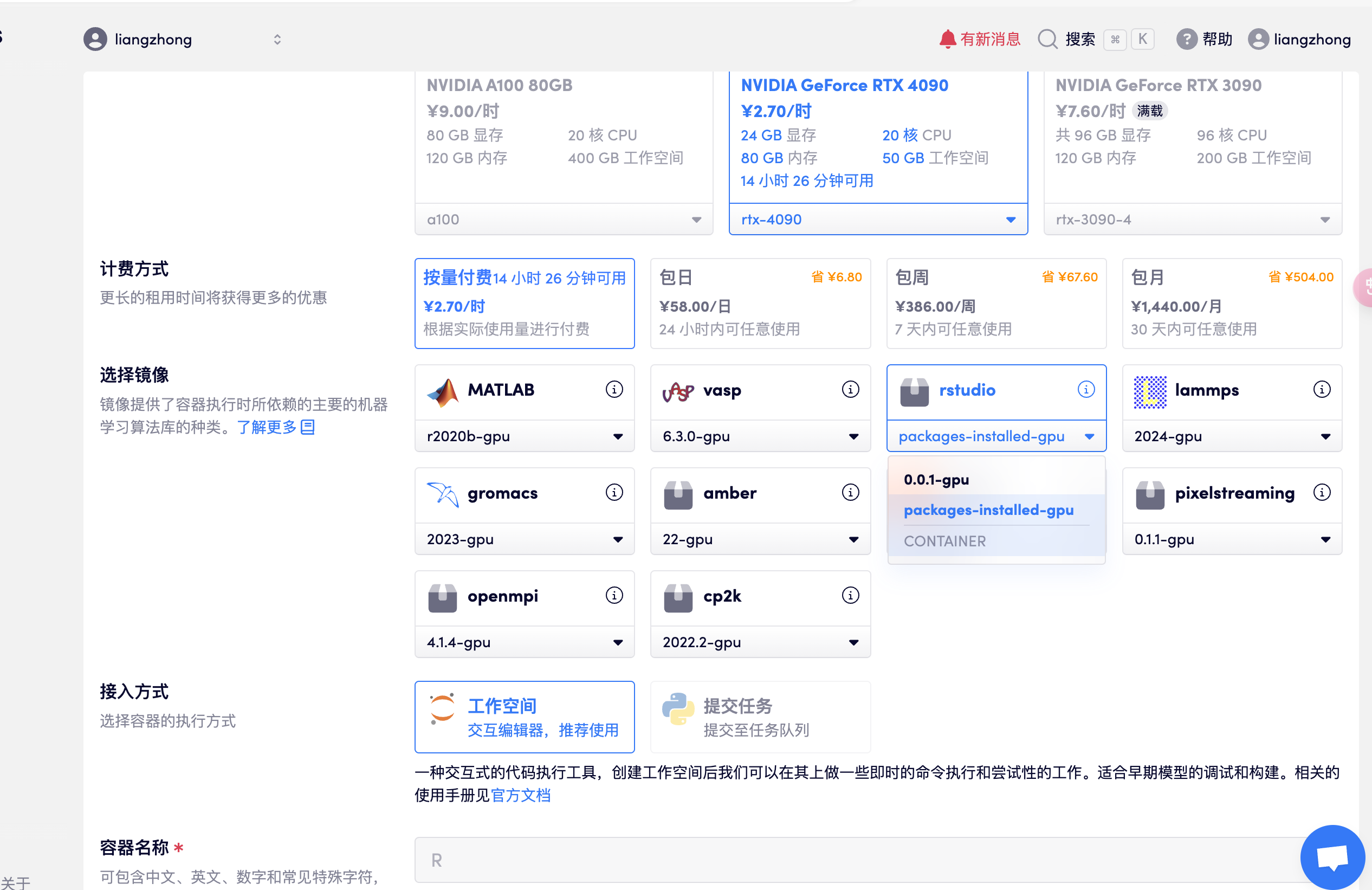
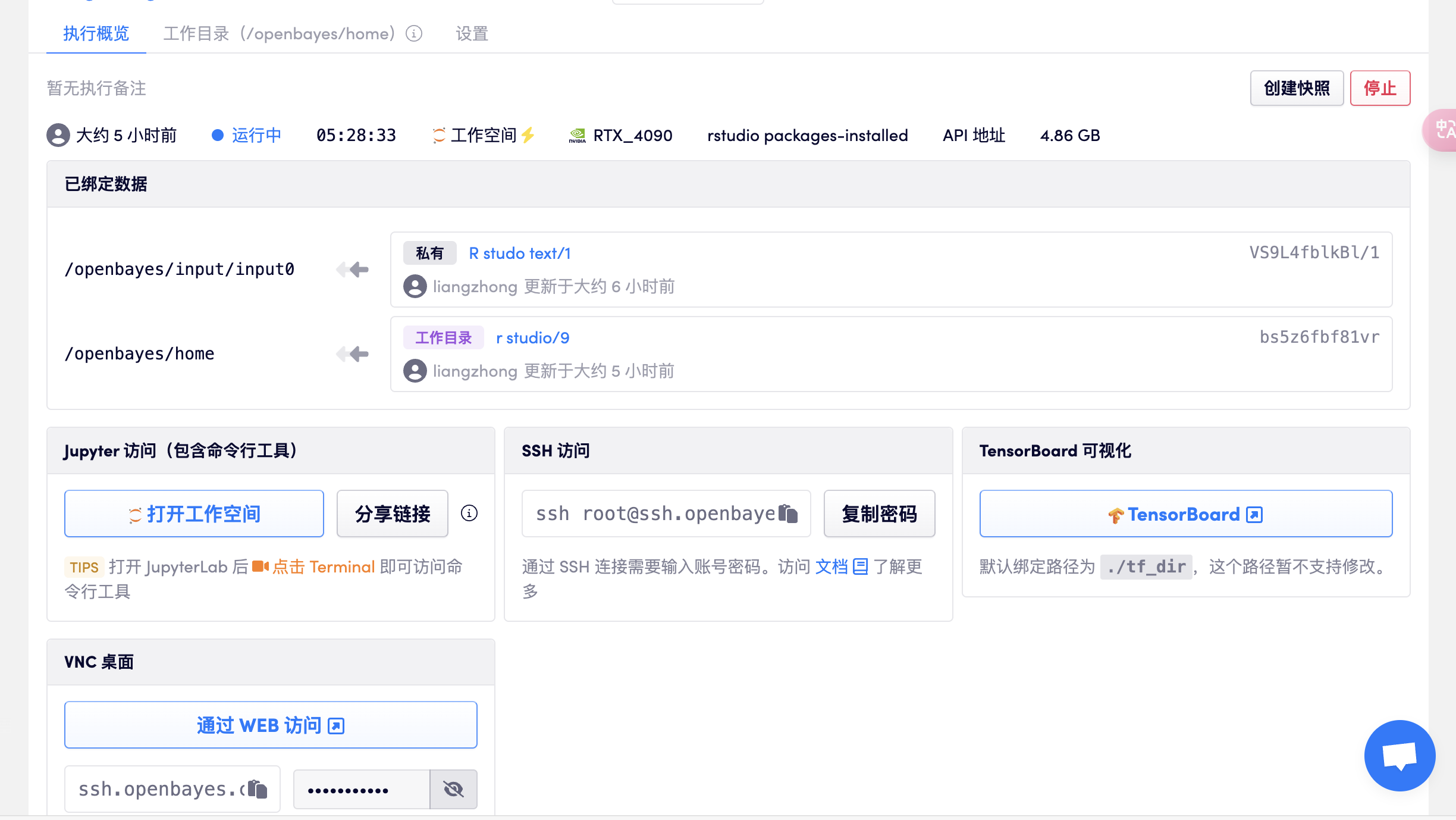
创建容器,打开 rstudio,选择 package-installed-gpu 版本,待资源分配后直接打开 API 地址进入 R studio(此步骤需要已完成实名认证),如下图所示:
- 注意:进入软件后,账号密码均为:rstudio
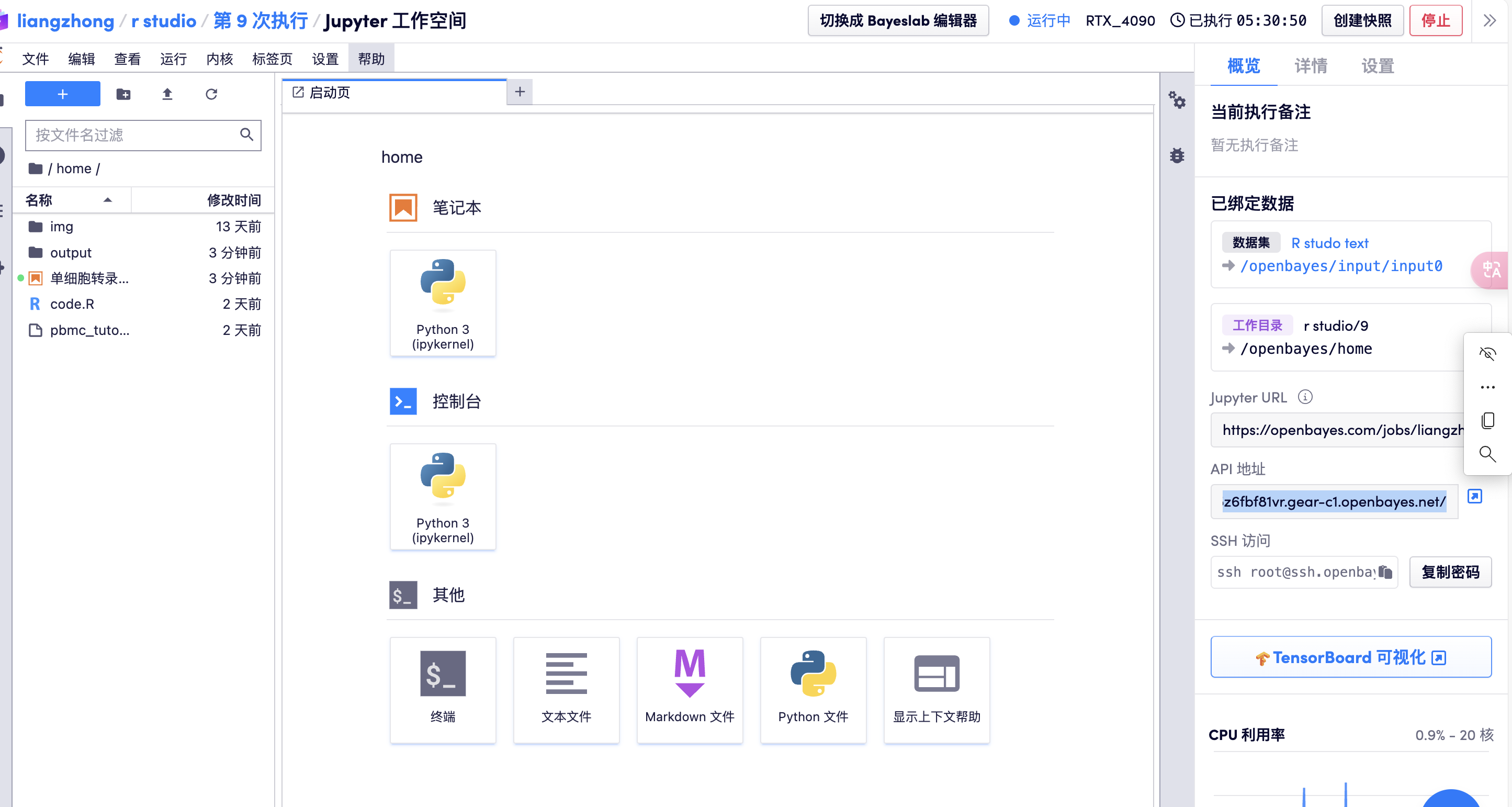
2. 上传数据
提前上传准备的数据(该数据均可在工作目录的 data 文件夹下进行下载使用):
(1) Illumina NextSeq 500 测序的单细胞的原始数据: barcodes.tsv.gz 、 features.tsv.gz 、 matrix.mtx.gz
(2) 细胞注释的 reference data: pbmc_multimodal.h5seurat 、 ref_Human_all.RData
#存放的目录如下
#此处可新建自定义文件夹,指定输入输出文件目录,本教程以 output 为例
rstudio@****(用户名)-bs5z6fbf81vrmain:~/home/output/data$ ls
barcodes.tsv.gz features.tsv.gz matrix.mtx.gz
rstudio@****(用户名)-bs5z6fbf81vr-main:~/home/output/reference_annotation$ ls
pbmc_multimodal.h5seurat ref_Human_all.RData下面在 R studio 中逐步按教程步骤输入代码。
#报错改为英文
Sys.setenv(LANGUAGE = "en")
options(stringsAsFactors = FALSE)
#清空所有数据
rm(list=ls())
set.seed(100)#!!!设置随机数,保证后续实验的可重复性一、设置 Seurat 对象
# I. Setup -----------------------------------------------------------------------
# ## a. Load libraries ----------------------------------------------------------------------
```{r libraries}
library(Seurat)
library(SeuratObject)
library(sctransform)
library(patchwork)
library(Seurat)
library(SeuratData)
library(patchwork)
library(SeuratData)
library(ggplot2)
library(tidyverse)
library(glmGamPoi)
library(tools)
library(dplyr)
library(patchwork)二、加载数据
# II. Load data-----------------------------------------------------------------------
### a. Load data-----------------------------------------------------------------
getwd()#查看当前工作目录
setwd("~/home/output/results")
#修改工作目录,此处可根据自定义文件夹,指定输入输出文件目录,本教程以 output 为例
pbmc.data <- Read10X(data.dir ="~/home/output/data")#设置 10x 原始数据的工作目录,读取
seurat <- CreateSeuratObject(counts = pbmc.data, project = "pbmc", min.cells = 3, min.features = 200)#创建 seurat 对象
seurat
An object of class Seurat
16356 features across 10066 samples within 1 assay
Active assay: RNA (16356 features, 0 variable features)
1 layer present: counts
#该样本有 10066 个细胞 ,10066 个单细胞的 RNA 表达数据,测序了 16356 个基因
#counts: 通常表示原始的基因表达计数数据
colnames([email protected])
[1] "orig.ident" "nCount_RNA" "nFeature_RNA" "percent.mt" "RNA_snn_res.0.1" "seurat_clusters"
[7] "celltype"
pbmc.data 数据如下图所示,reads 数,行名为基因,列名为细胞 barcodes
pbmc.data[c(“CD3D”, “CD8A”, “PTPRC”), 1:30] 3 x 30 sparse Matrix of class “dgCMatrix” [[ suppressing 30 column names ‘AAACCTGAGCGAAGGG-1’, ‘AAACCTGAGGCCCTTG-1’, ‘AAACCTGAGGGTTTCT-1’ … ]]
CD3D 2 . . 3 6 4 1 1 3 . 6 3 4 2 1 2 1 . . 2 2 5 2 3 . 6 . . 4 3 CD8A . . . . 5 . . . 1 . . . . . . . . . . . . . 1 1 . 6 . . . 6 PTPRC 1 2 2 2 3 5 3 3 3 1 3 . 2 4 1 . 1 . 2 9 . 3 4 . 1 2 . 2 1 4
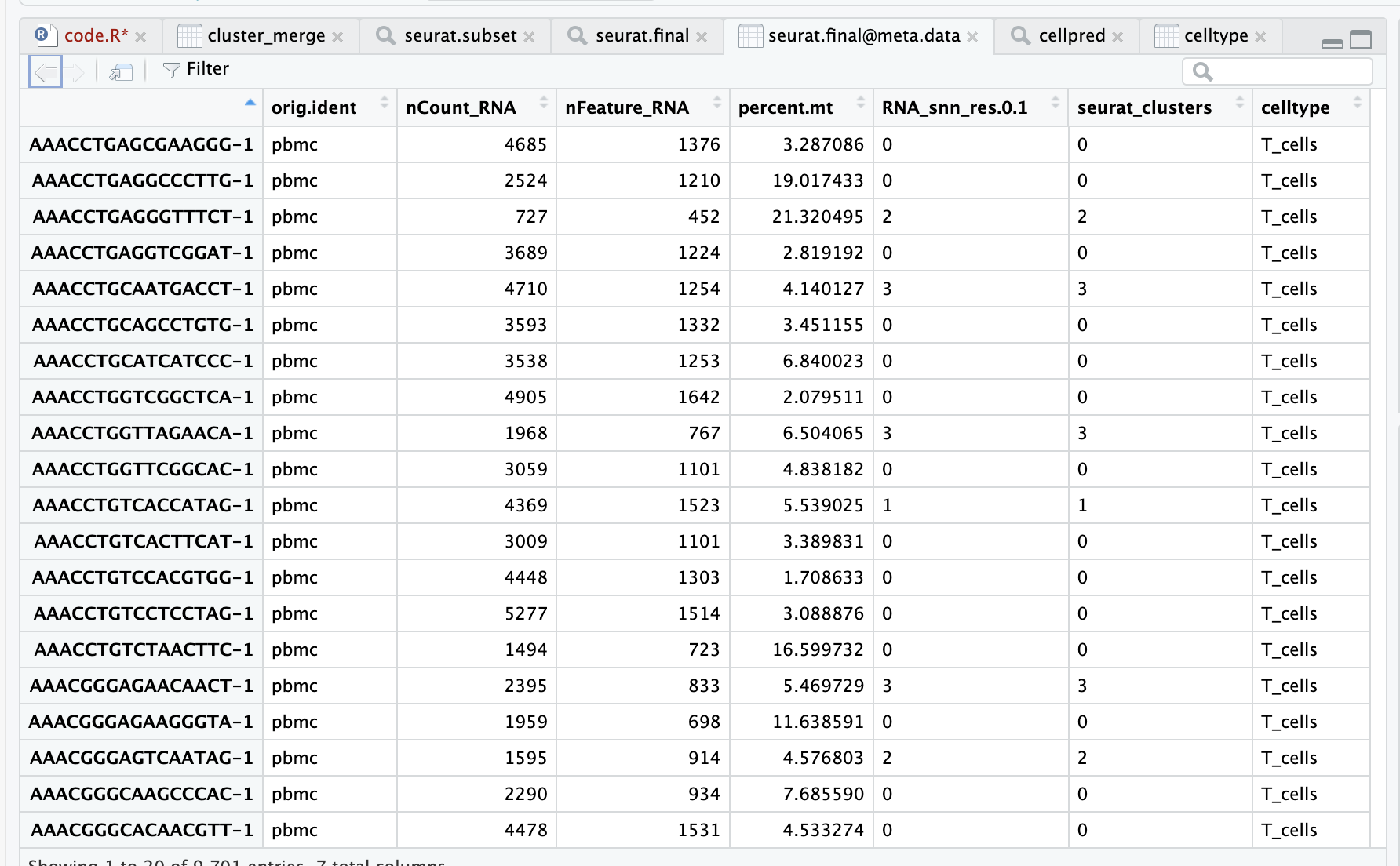
- 表格信息说明:
- 行名为 barcodes
- orig.ident 是样本信息,
- nCount_RNA 是总基因表达
- nFeature_RNA 是基因数量
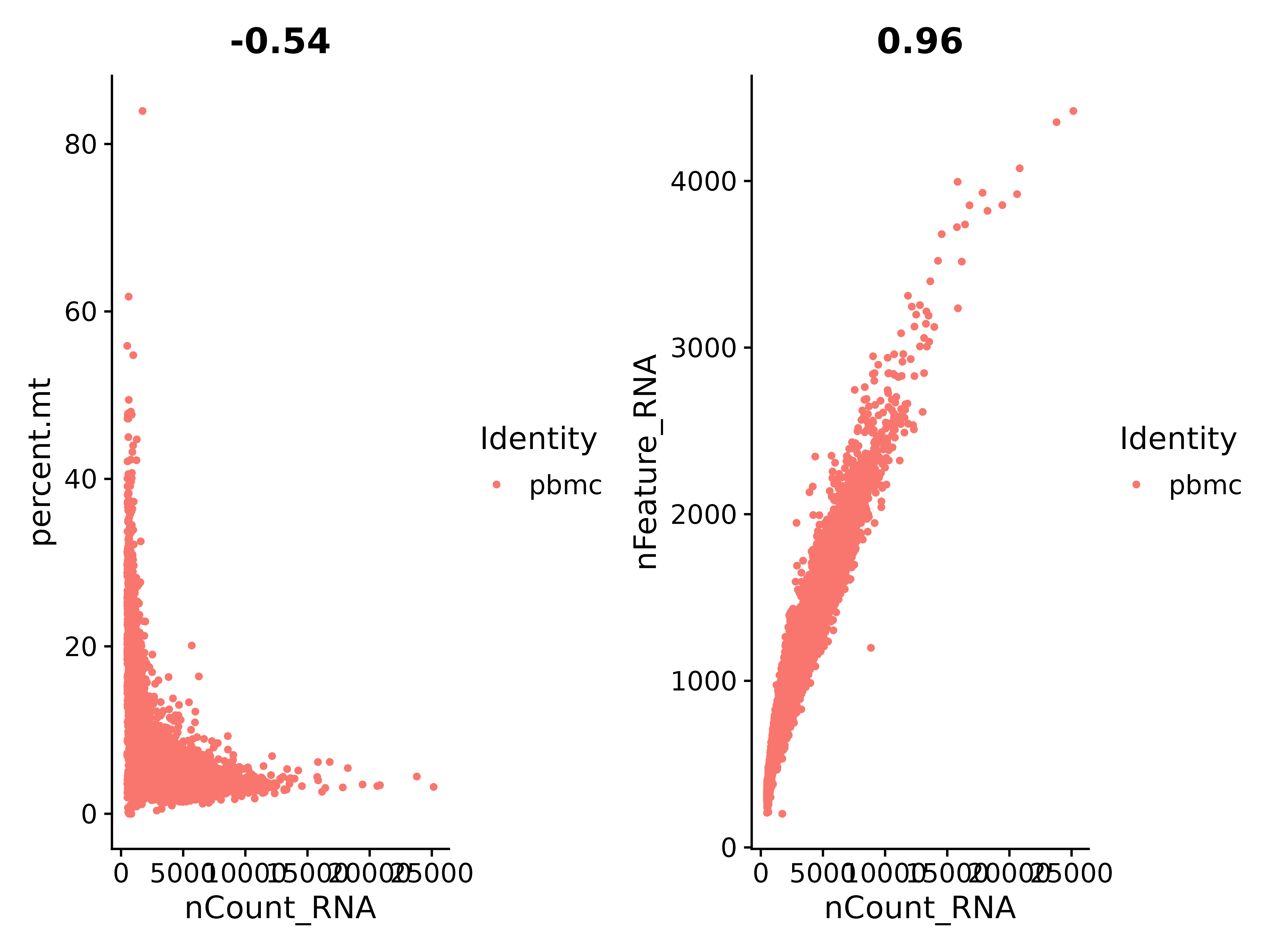
根据上图,我们查看一下 nCount_RNA 和 percent.mt(线粒体基因)的比例关系:
可以看到 nCount_RNA 和 nFeature_RNA 正比例,系数为 0.96,相关性很好
tips:MT- 开头的所有基因是线粒体基因。
三、标准预处理
以下步骤涵盖了 Seurat 中 scRNA-seq 数据的标准预处理工作流程。这些流程包括基于 QC 指标选择和过滤细胞、数据标准化和缩放以及检测高度可变的特征。
Seurat 可以查看 QC 指标并根据任何用户定义的标准过滤单元格。
# III. Pre-processing----------------------------------------------------------------
### a. Plots for percent mito, nFeature---------------------------------------------
```{r preprocessing}
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
seurat <-PercentageFeatureSet(seurat, pattern = "^MT-", col.name = "percent.mt")
# Use FeatureScatter is typically used to visualize feature-feature relationships, but can be used
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt",)
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
ggsave("质控_1.png", plot1 + plot2, width = 8, height = 6, dpi=600)
#plot3 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.ribo")
#plot3
下面进行可视化 QC 指标,并使用这些指标来过滤单元格。
看线粒体基因比例,使用小提琴图可视化 QC 指标
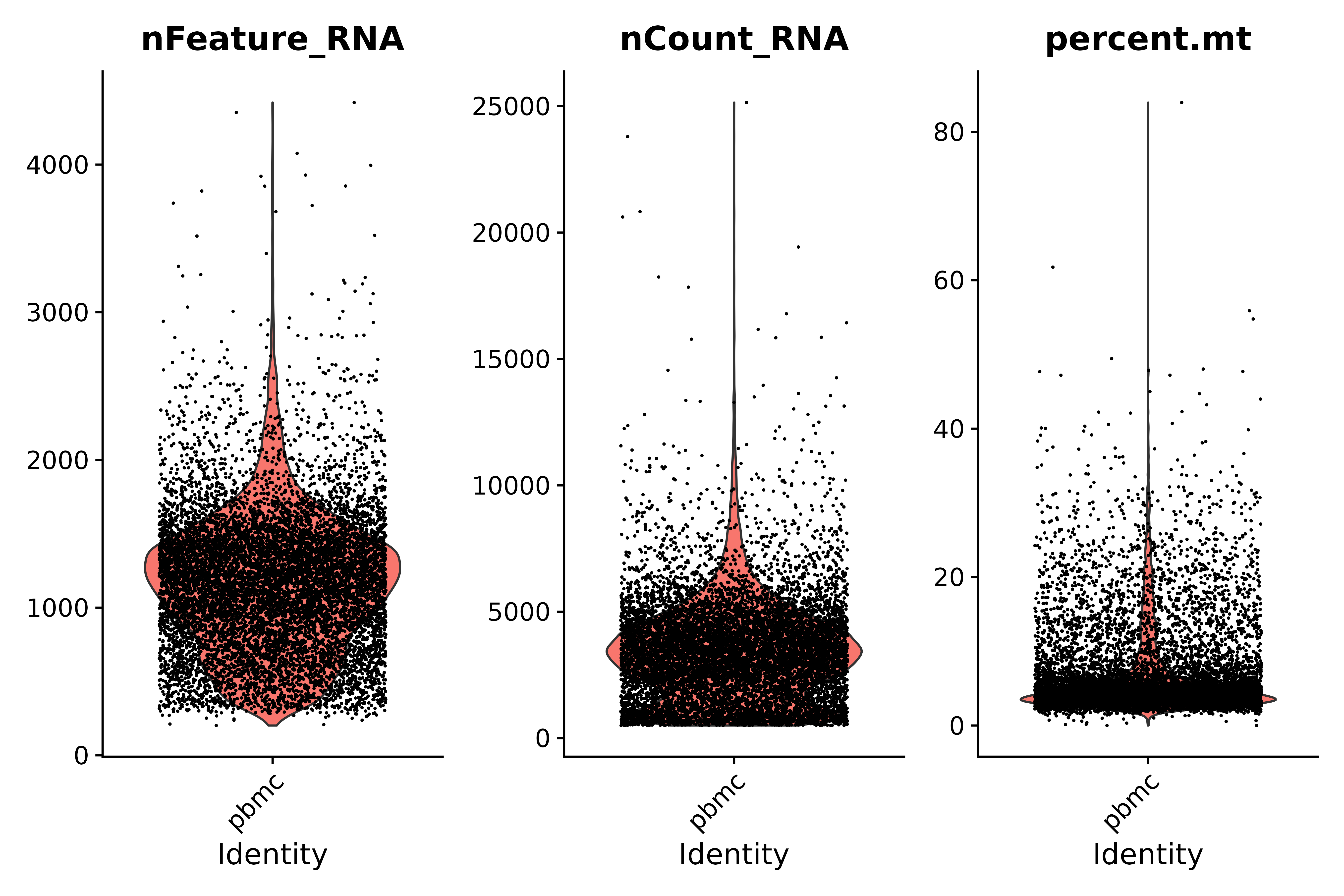
- 质控说明:
- 低质量细胞或空液滴通常基因很少,nFeature_RNA > 200 & nCount_RNA > 500
- 细胞双倍体或多倍体可能表现出异常高的基因计数,根据我们的图,nFeature_RNA < 2500 & nCount_RNA > 500
- 低质量/濒死细胞通常表现出广泛的线粒体很高,percent.mt < 25
# Visualize QC metrics as a violin plot
vln1 <- VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"#,"percent.ribo"
),ncol=3)
vln1
ggsave("质控_2.png", vln1, width = 9, height = 6, dpi=600)seurat.subset <- subset(seurat, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & nCount_RNA > 500 & nCount_RNA < 10000 &percent.mt < 25)
vln3 <- VlnPlot(seurat.subset, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
vln3
ggsave("质控_3.png", vln3, width = 9, height = 6, dpi=600)
pdf("qc_plots.pdf");plot1 + plot2;vln1;vln3;dev.off()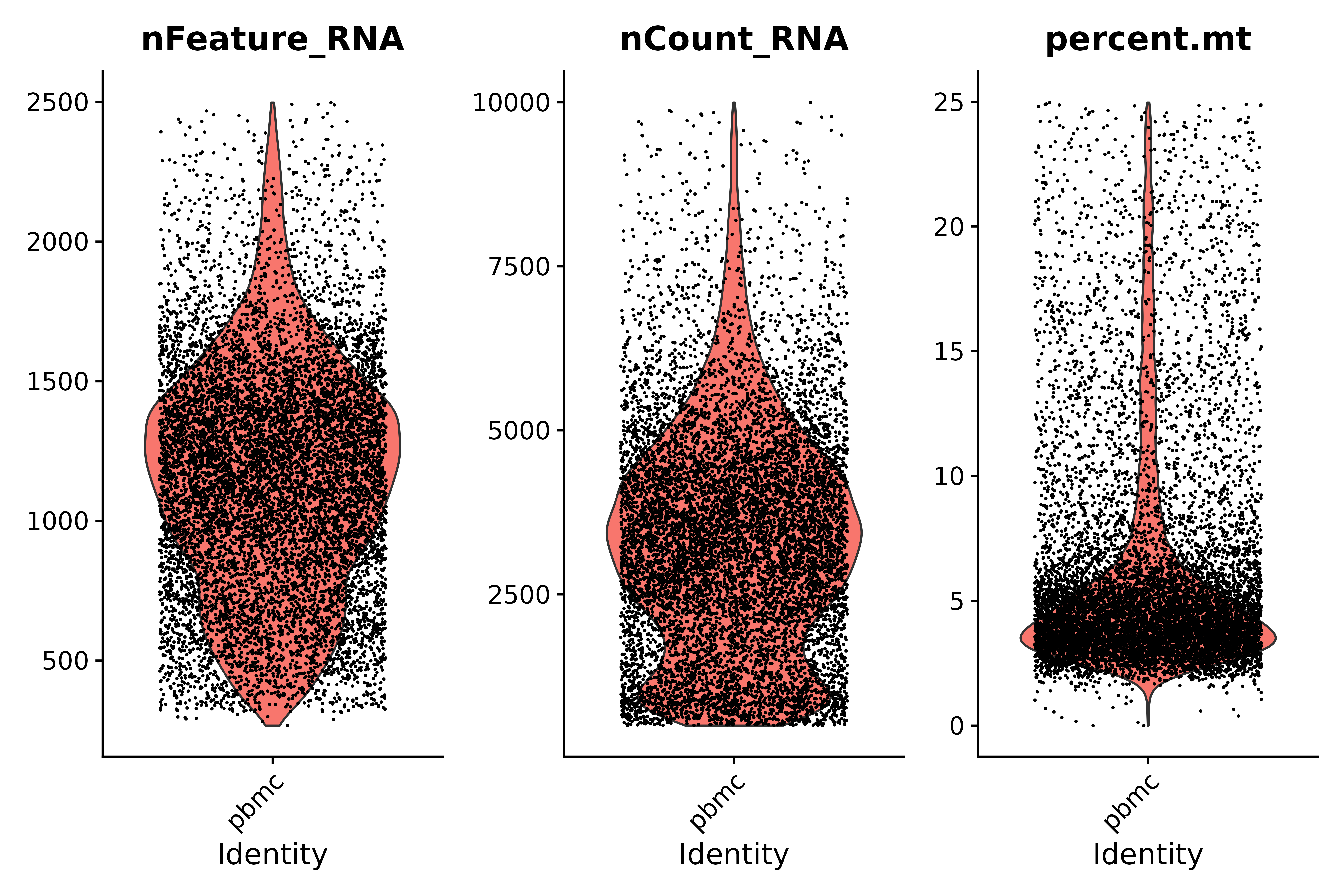
四、规范化数据并识别高度可变的特征(特征选择)
在单细胞转录组数据分析中,我们首先进行数据归一化处理,以确保不同细胞间的基因表达水平可以进行有效比较。
1. 数据归一化处理
- 数据归一化:我们首先将每个细胞中特定基因的表达量 (count) 除以该细胞的总表达量 (total count),这一步骤是为了消除细胞间测序深度的差异,使得不同细胞的基因表达水平可以放在相同的基准上进行比较。
- 缩放调整:归一化后的表达量接着乘以一个缩放因子,这里是 10000,这样做是为了将基因表达值调整到一个更合适的量级,便于后续分析。
- 对数转换:最后,我们对调整后的表达量进行对数转换,这有助于进一步稳定方差,使得高表达和低表达基因的差异更加明显,为后续的分析步骤打下基础。
- 标准化:通过上述步骤,我们将基因表达值标准化到每 10000 个 UMI 的水平,这有助于消除细胞间测序深度差异的影响。
2. 接下来,我们进行变异特征的寻找:
在归一化和标准化之后,我们通过分析基因表达的变异性来识别高变基因。这些基因在不同细胞中的表达水平有显著差异,可能在某些细胞中高度表达,而在其他细胞中表达水平较低。
# IV. Normalize Data----------------------------------------------------------------
seurat.subset <- NormalizeData(seurat.subset, normalization.method = "LogNormalize", scale.factor = 10000)
seurat.subset <- FindVariableFeatures(seurat.subset, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(seurat.subset), 10)
# plot variable features with and without labels
plot3 <- VariableFeaturePlot(seurat.subset)
plot4 <- LabelPoints(plot = plot3, points = top10, repel = TRUE)
ggsave("VariableFeaturePlot_4.png", plot3 + plot4, width = 12, height = 6, dpi=600)
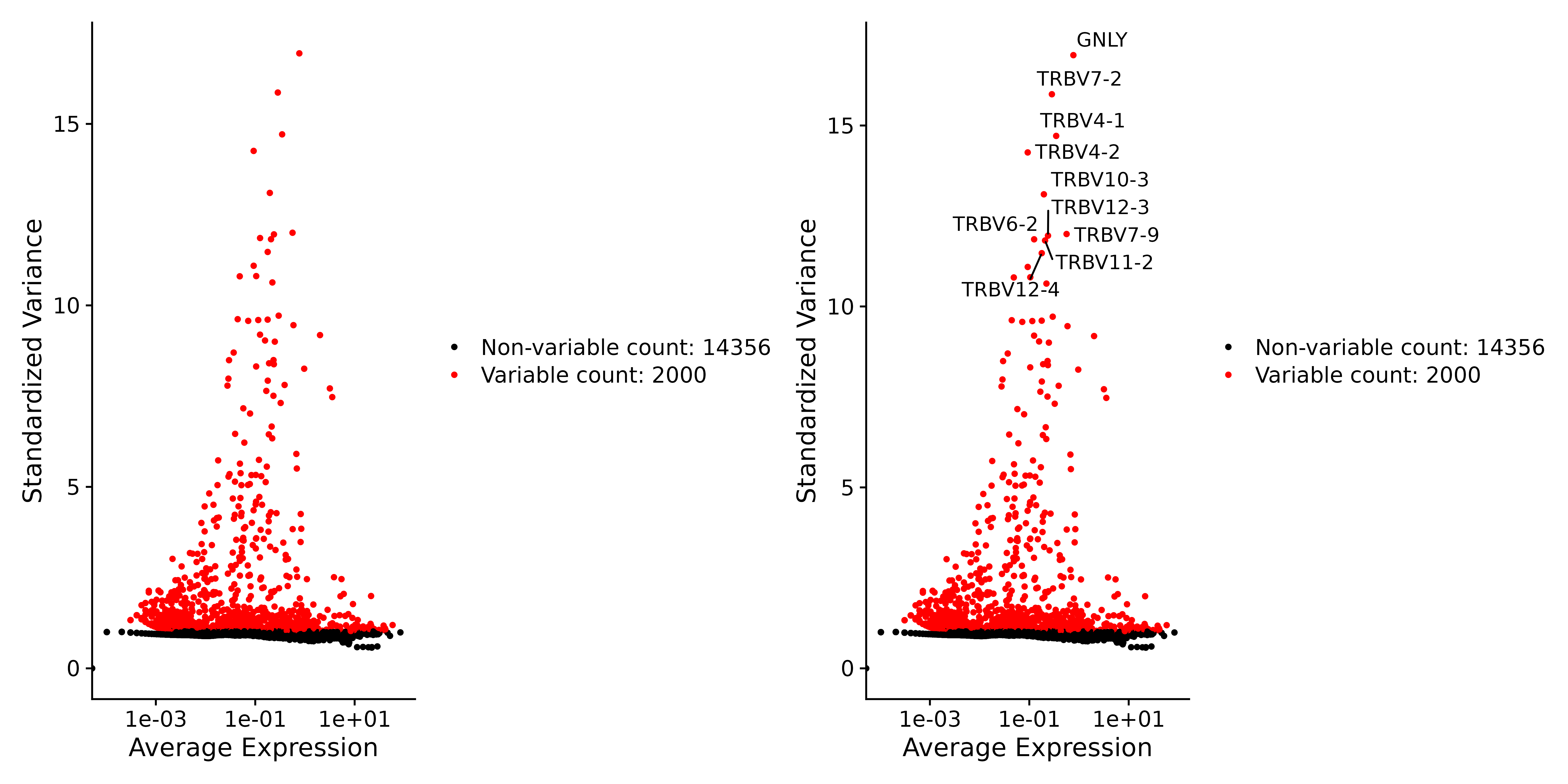
五、缩放数据 (Scale Data) 并运行主成分分析 (Run PCA)
在单细胞转录组数据分析中,通过数据缩放和 PCA 可以有效地减少数据的维度,提取细胞间变异的主要来源,为理解细胞的异质性和功能提供重要信息。
1. 数据缩放 (Scale Data):数据缩放是一种线性变换,目的是将基因表达数据转换成标准正态分布,即数据的均值 (mean) 为 0,标准差 (standard deviation) 为 1 。这种转换有助于减少不同基因表达量级之间的差异,使得数据更加规范化。通过缩放,可以降低数据中的极端值或异常值的影响,使得数据分布更加均匀,为后续的分析步骤提供更稳定的输入。这一步也是为 PCA(主成分分析)做准备,因为 PCA 在处理标准化的数据上执行效果更佳。
2. 运行主成分分析 (Run PCA):
- PCA 是一种统计方法,它通过正交轴变换将数据转换到新的坐标系统中,使得数据的任何投影的第一大方差在第一个坐标(称为第一主成分)上,第二大方差在第二个坐标上,依此类推。
- 在单细胞转录组分析中,PCA 用于根据基因表达量的差异将每个细胞分开。每个细胞可以被视为高维空间中的一个点,而 PCA 则将这些点在低维空间中重新表示,同时保留尽可能多的变异信息。
- 每个主成分(PCA)本质上代表了细胞的一个「元特征」,即细胞的一种主要变异方向。在 PCA 结果中,越靠前的主成分,其在数据变异中的权重越大,意味着它包含了更多的信息。
- 通过 PCA,可以将细胞的高维表达信息压缩成几个关键的主成分,这些主成分可以用于后续的聚类分析、可视化或其他下游分析。
all.genes <- rownames(seurat.subset)
seurat.subset <- ScaleData(seurat.subset, features = all.genes)
seurat.subset <- RunPCA(seurat.subset, features = VariableFeatures(object = seurat))##### Examine and visualize PCA results a few different ways-------------
print(seurat.subset[["pca"]], dims = 1:5, nfeatures = 5)
p1 <- VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
p1
ggsave("VizDimLoadings_5.png", p1, width = 9, height = 6, dpi=600)
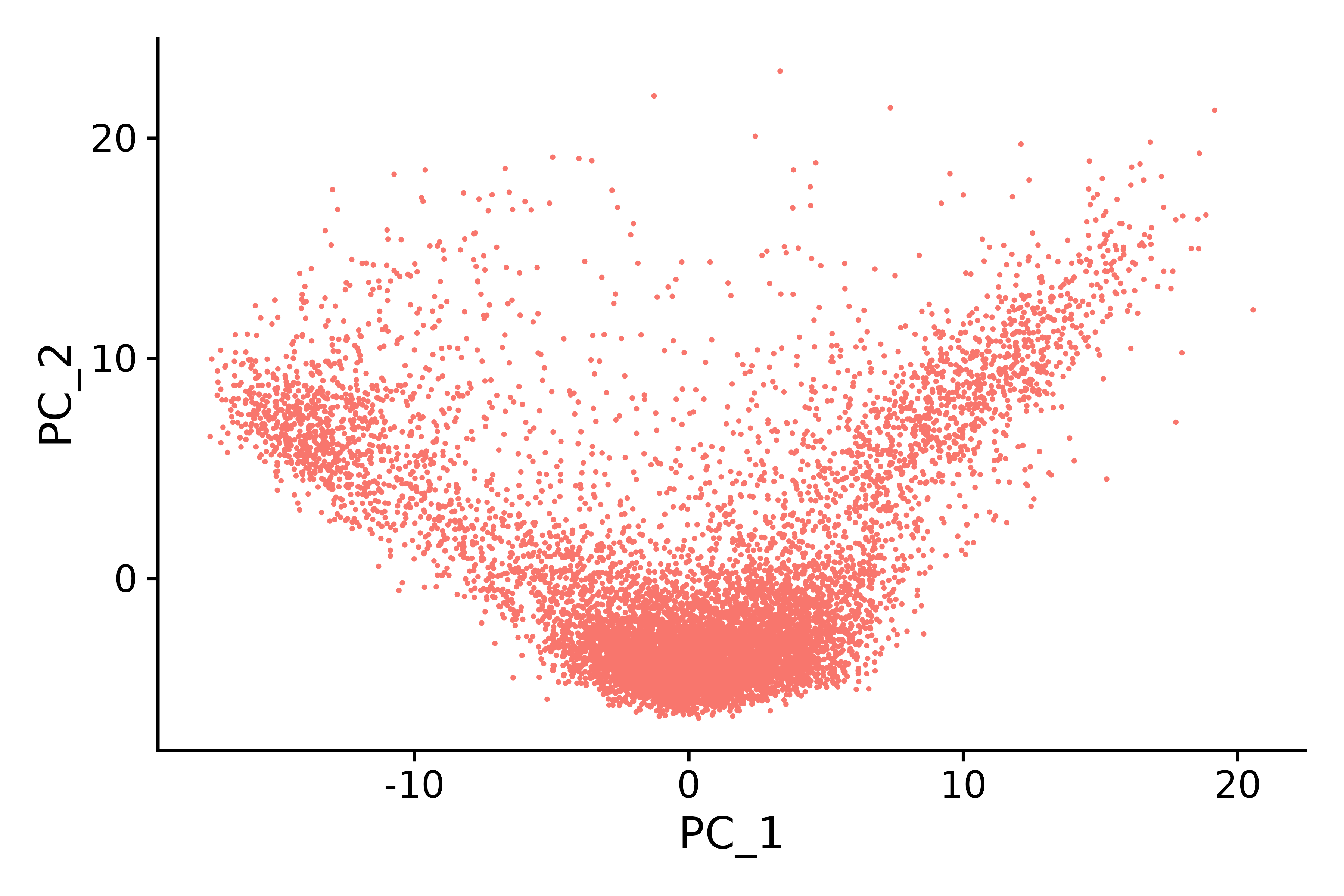
p2 <- DimPlot(seurat.subset, reduction = "pca") + NoLegend()
p2
ggsave("pca_6.png", p2, width = 6, height = 4, dpi=600)
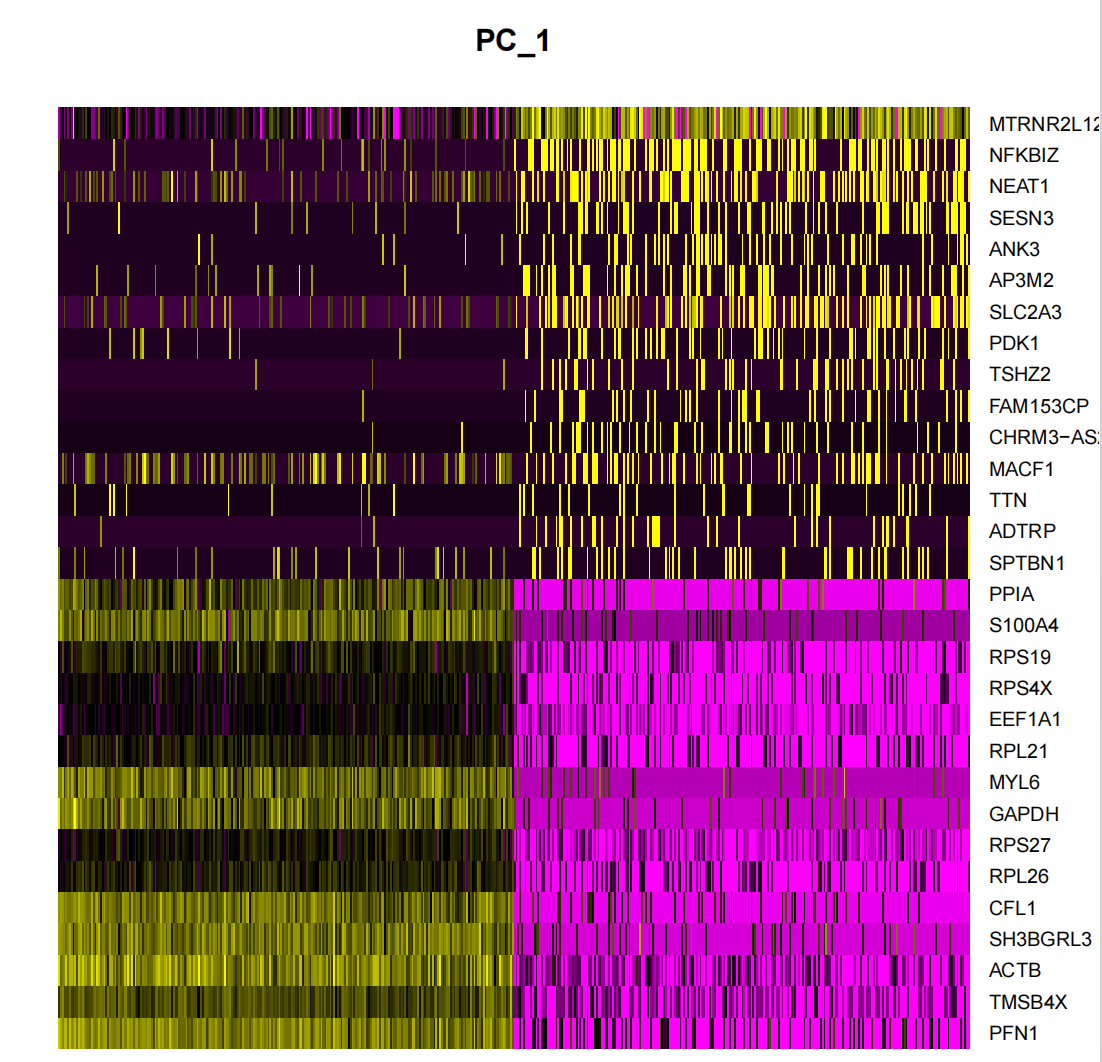
p3 <- DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
p3
ggsave("pca_1dims.png", p3, width = 6, height = 4, dpi=600)
p4 <- DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
p4
ggsave("pca_15dims.png", p4, width = 10, height = 10, dpi=600)
p5 <- ElbowPlot(seurat.subset)
p5
ggsave("ElbowPlot.png", p5, width = 10, height = 10, dpi=600)
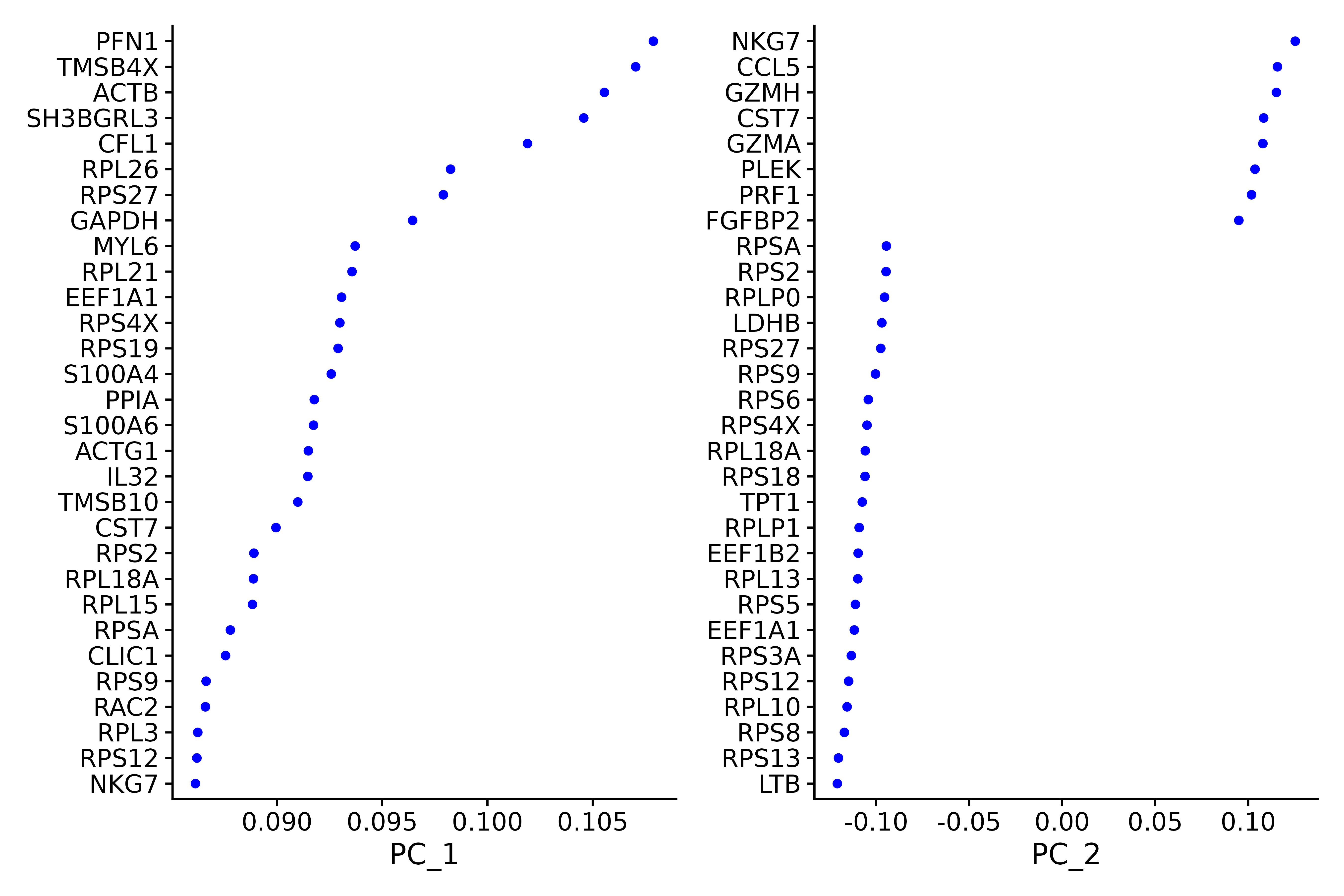
DimHeatmap() 是一款强大的数据分析工具,它使得研究者能够轻松地识别和探索数据集中异质性的主要来源。在进行主成分分析 (PCA) 之后,DimHeatmap() 特别有助于确定哪些主成分 (PCs) 应该被纳入,以便为后续的深入分析提供信息。这一过程涉及到根据 PCA 得分对细胞和特征进行排序,确保了分析的精确性和相关性。
此外,DimHeatmap() 通过允许用户设定一个特定的细胞数量,专门在数据谱的两端绘制那些表现出极端特征的「极端」细胞。这种方法显著提升了在处理大型数据集时的绘图效率,因为它专注于数据中最为显著的部分,从而加快了整个可视化过程。

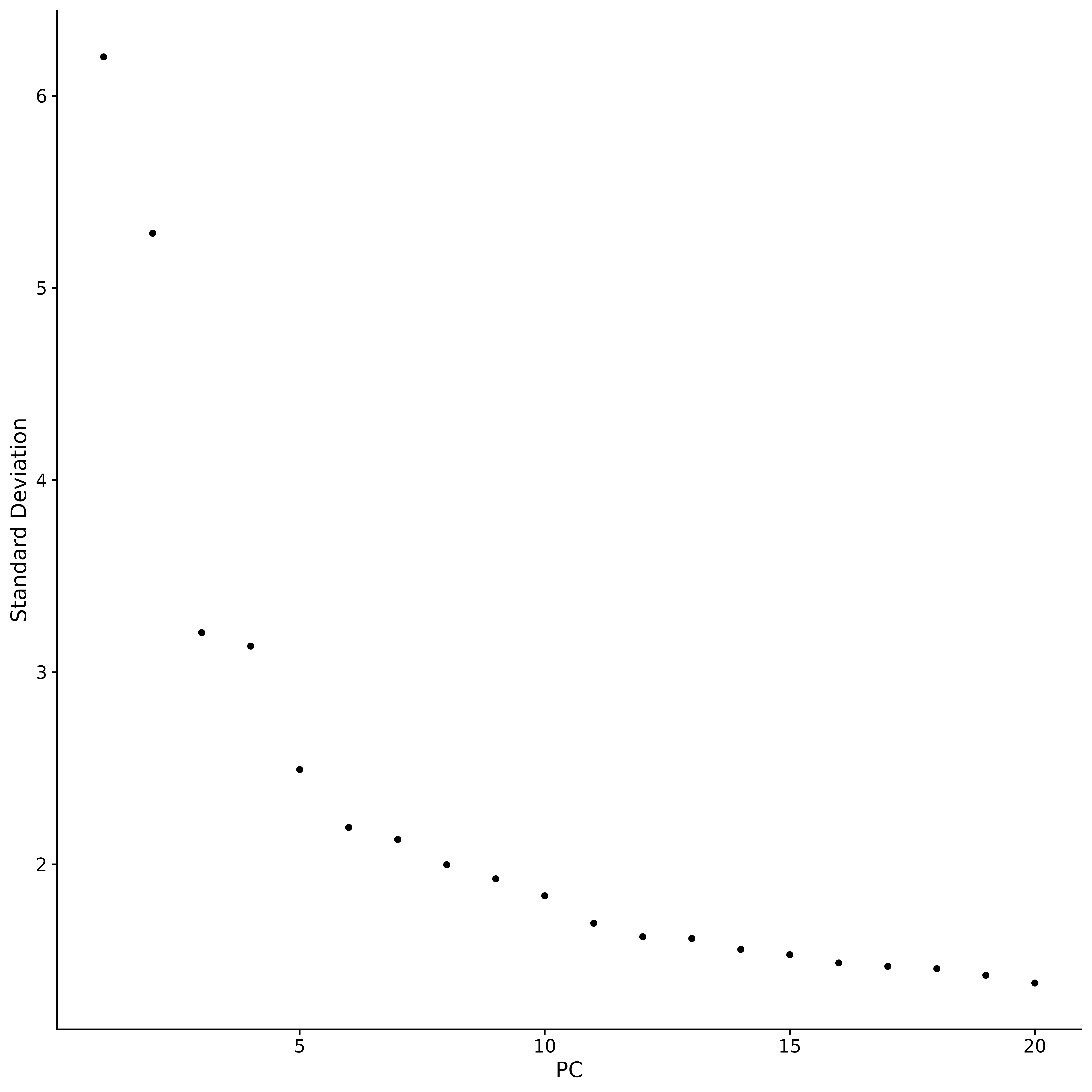
通过「肘形图」来确定保留多少个主成分进行后续分析是主成分分析 (PCA) 中的一个常见做法。该图表明大多数真实信号被捕获在前 17 个主成分中,在肘形图中,我们寻找一个转折点,即「肘点」。在这个点之前,每增加一个主成分都会显著提高数据的解释度,但在这个点之后,增加主成分所带来的增益开始递减,这表明已经达到了一个平衡状态。 基于肘形图的分析结果,我们选取前 17 个主成分进行后续分析。 因为增加更多的主成分并不会显著提高数据的区分度,但会导致计算量的不必要增加。如果继续增加主成分的数量,虽然可能会轻微提高数据的区分度,但这种提升并不显著,而且会导致计算成本显著上升。因此,选择 17 个主成分是一个平衡了区分度和计算效率的合理选择。
pdf("dimreduction_plots.pdf")#保存为 pdf 文件
VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
DimPlot(seurat.subset, reduction = "pca")
DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
ElbowPlot(seurat.subset)
dev.off()seurat.subset.dimReduction <- seurat.subset
seurat.subset.dimReduction <- FindNeighbors(seurat.subset.dimReduction, reduction="pca", dims = 1:17)
saveRDS(seurat.subset.dimReduction, file = "~/home/output/data/seurat_pre_FindClusters.rds")
# seurat.subset.dimReduction <- readRDS("~/home/output/data/seurat_pre_FindClusters.rds")六、将细胞聚类
### 3. Find clusters,聚类
```{r find_clusters}
seurat.final <- FindClusters(object = seurat.subset.dimReduction, resolution = 0.1)
head(Idents(seurat.final), 5)
seurat.final <- RunUMAP(seurat.final, reduction="pca", dims=1:17)
#Visualize with UMAP
p1<- DimPlot(seurat.final, reduction = "umap")
p1
ggsave("find_clusters.png", p1, width = 6, height =5 , dpi=600)
saveRDS(seurat.final, file = "~/home/output/data/seurat_final.rds")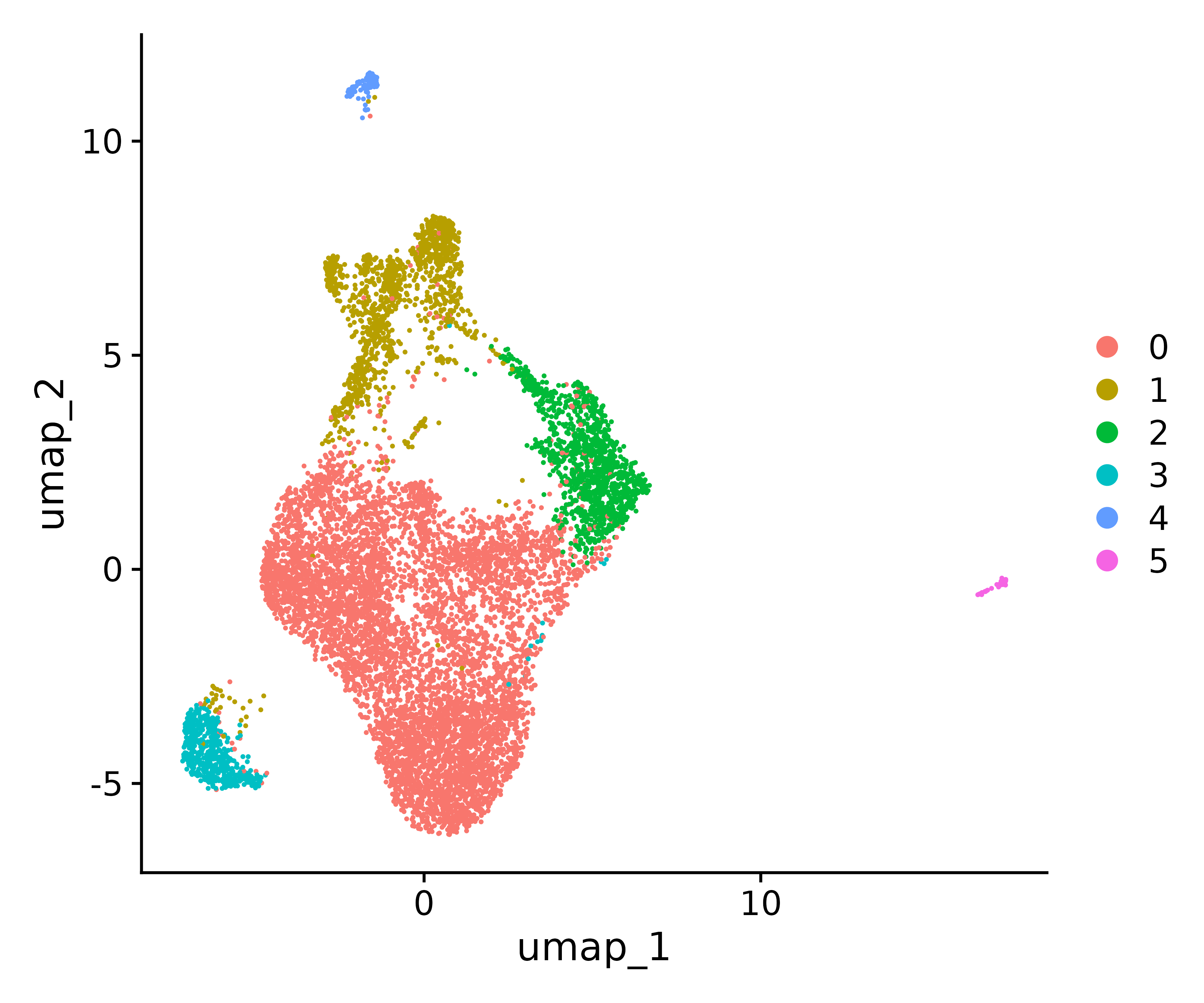
可视化
查看基因在不同的 cluster 表达的 3 种常见的图的绘制有:
1. 热图 2. 气泡图 3.featureplot
table(Idents(seurat.final))
# find markers between cluster1 and cluster 2
##用来找 cluster 之间的寻找差异表达特征
cluster2.markers <- FindMarkers(seurat.final, ident.1 = 1,ident.2 = 2)
head(cluster2.markers, n = 5)
# find markers for every cluster compared to all remaining cells, report only the positive ones
#找每个 cluster 的 marker 基因
pbmc.markers <- FindAllMarkers(seurat.final, only.pos = TRUE)
ClusterMarker_noRibo <- pbmc.markers[!grepl("^RP[SL]", pbmc.markers$gene, ignore.case = F),]
ClusterMarker_noRibo_noMito <- ClusterMarker_noRibo[!grepl("^MT-", ClusterMarker_noRibo$gene, ignore.case = F),]
###去除核糖体基因和线粒体基因的影响
ClusterMarker_noRibo_noMito %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
###展示每个 cluster 的 top5 的基因
heatmap <- DoHeatmap(seurat.final, features = top5$gene) + NoLegend()
ggsave("heatmap.png", heatmap, width = 10, height =5 , dpi=600) 该图为热图展示 marker 基因
该图为热图展示 marker 基因
g =unique(top5$gene)
##定义 top5 的 gene 列,作为基因集 g,也可以自定义
p <- DotPlot(seurat.final,features=g)+
coord_flip() + # 翻转坐标轴
theme(
panel.grid = element_blank(), # 移除网格线
axis.text.x = element_text(angle = 0, hjust = 0.5, vjust = 0.5) # 旋转 x 轴标签
)
ggsave("allmarker_dotpot.png", p, width = 8, height =7 , dpi=600)
#气泡图展示 marker 基因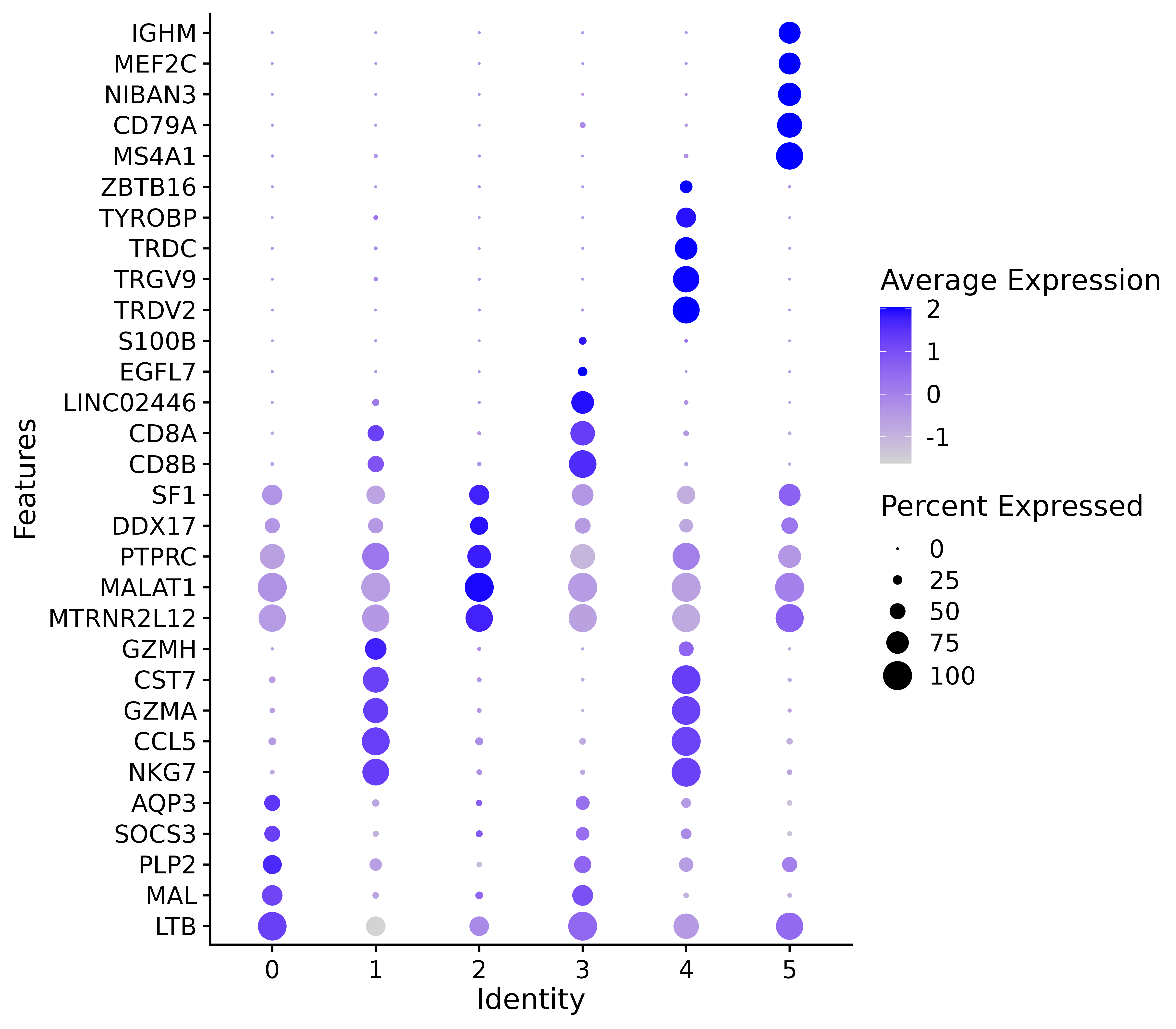
该图为气泡图展示 marker 基因
# 加载 dplyr 包
library(dplyr)
# 提取 cluster 和 gene 列
cluster_gene_mapping <- top5 %>%
select(cluster, gene)
# 查看结果
head(cluster_gene_mapping,30)
write.csv(cluster_gene_mapping,"cluster_allmarkers_TOP5.csv", row.names = p <- FeaturePlot(seurat.final, features = g)
ggsave("allmarker_featurepot.png", p, width = 15, height =15 , dpi=600)
###查看自定义感兴趣的基因
p2 <- FeaturePlot(seurat.final, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
ggsave("allmarker_featurepot2.png", p2, width = 15, height =15 , dpi=600)


七、数据分析
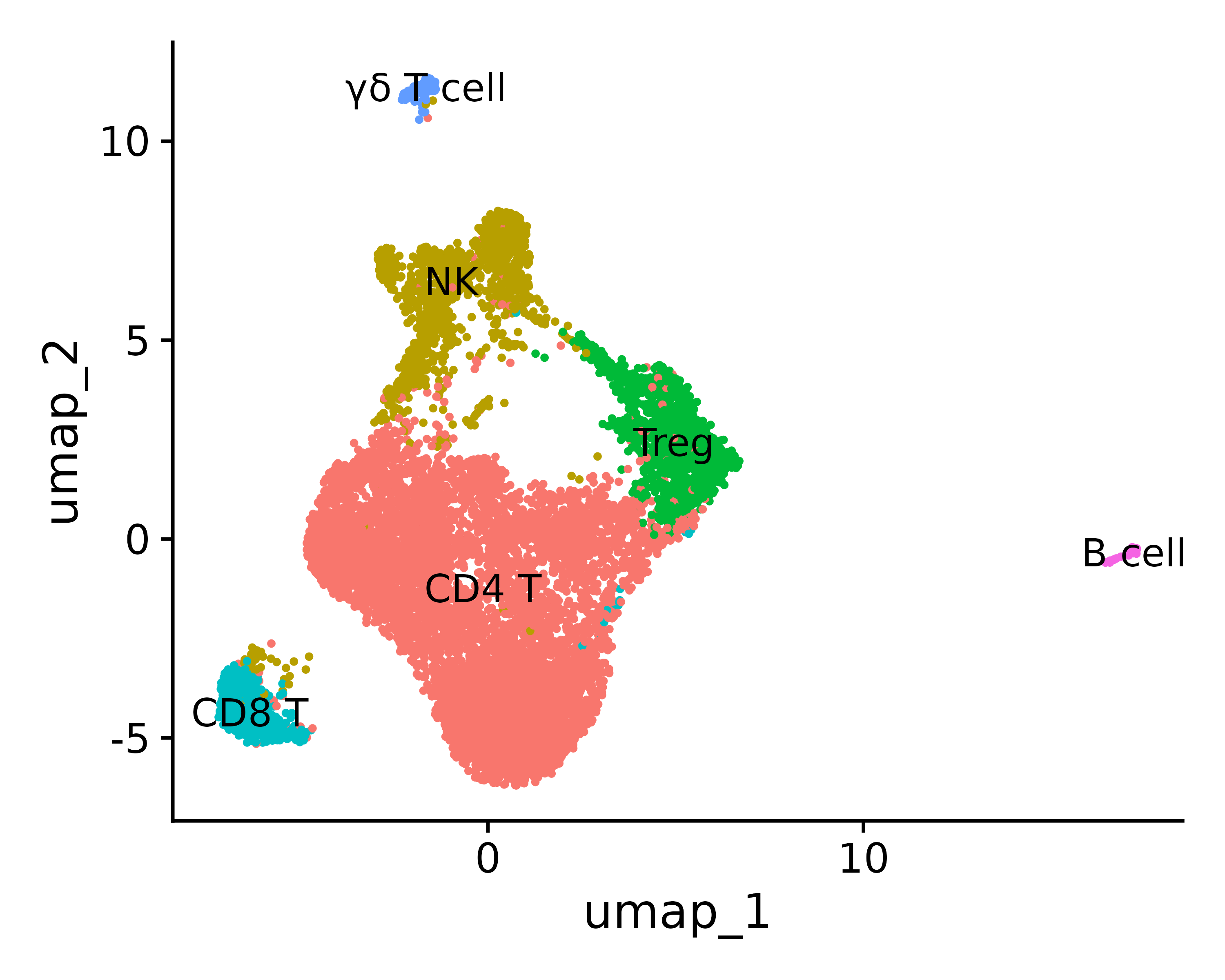
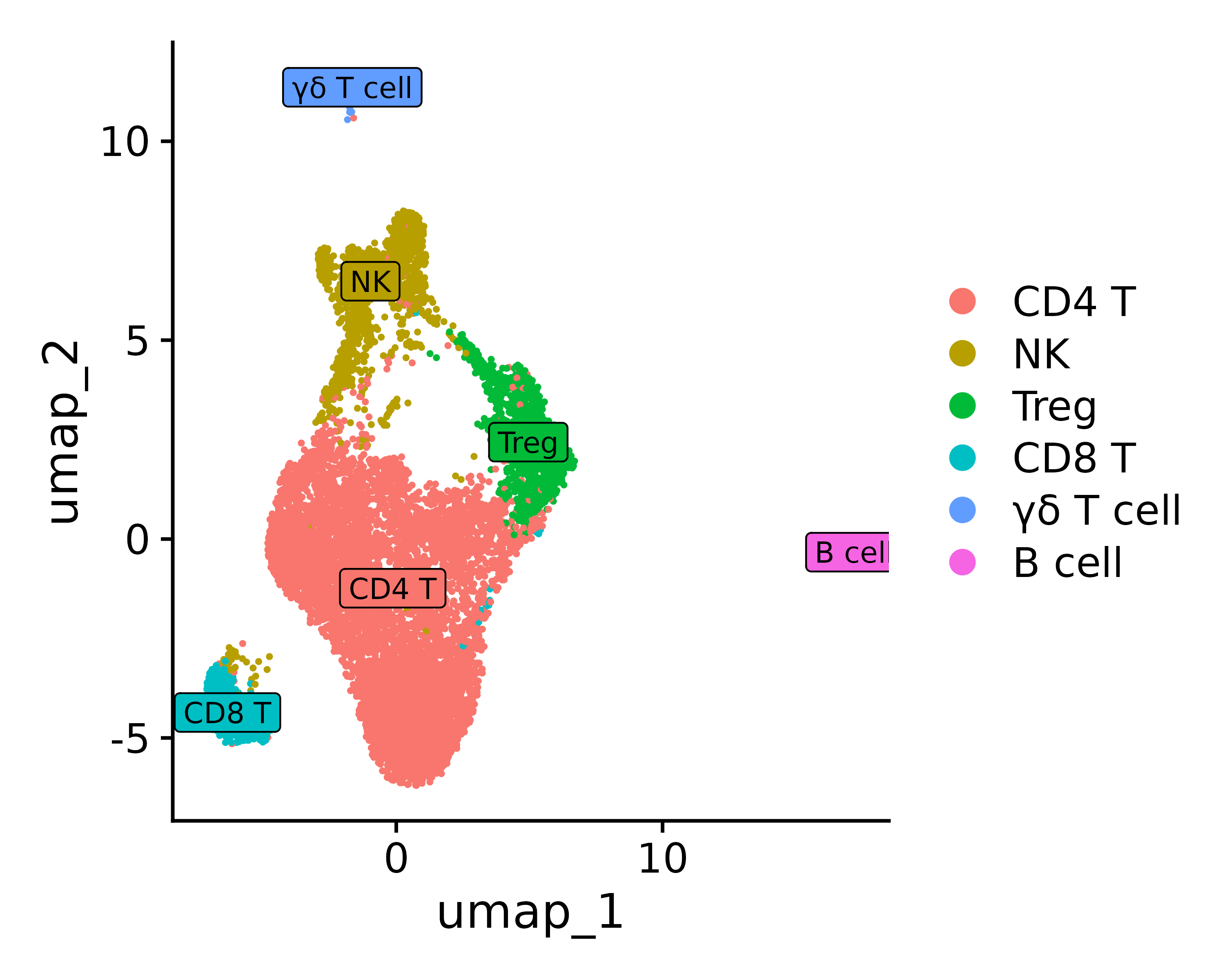
本步骤介绍在单细胞转录组数据分析中,如何利用特定的标记基因 (marker genes) 来确定和定义细胞类型的过程。
假设你根据 top 的 marker 基因等方法确定了细胞类型,可以定义身份类型
0 1 2 3 4 5 “CD4 T”, “NK”, “Treg”, “CD8 T”, “γδ T cell”, “B cell”
(以下注释只是参考示范,并不是实际注释结果)
new.cluster.ids <- c("CD4 T", "NK", "Treg", "CD8 T", "γδ T cell",
"B cell")
names(new.cluster.ids) <- levels(seurat.final)
seurat.final <- RenameIdents(seurat.final, new.cluster.ids)
p <- DimPlot(seurat.final, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
p3 = DimPlot(seurat.final, reduction = "umap", label = TRUE, label.size = 3, pt.size = .3, label.box = T)
ggsave("annotation_umap.png", p, width = 5, height =4 , dpi=600)
ggsave("annotation_umap2.png", p3, width = 5, height =4 , dpi=600)