Command Palette
Search for a command to run...
AlphaFold 应用新里程碑!剑桥大学团队提出 AlphaFold-Metainference,精准预测无序蛋白质结构集合
自 2018 年底 AlphaFold 横空出世以来,蛋白质结构预测领域在 AI 的加持下可谓是发生了翻天覆地的变化。如今,AlphaFold 不仅在预测精度方面令人刮目相看,更是在最新的迭代中将预测范围逐步拓宽。也难怪中科院院士施一公曾在媒体面前毫不吝啬对其评价道,「依我之见,这是人工智能对科学领域最大的一次贡献,也是人类在 21 世纪取得的最重要的科学突破之一,是人类在认识自然界的科学探索征程中一个非常了不起的历史性成就」。
尽管 AlphaFold 引领的蛋白质结构预测革命如此迅速,也仍有一些悬而未决的问题拦路在前。其中,针对无序蛋白质的研究在生命科学领域中一直是个难题。这些蛋白质在细胞信号传导、调控过程以及多种疾病中发挥着关键作用,但由于它们在结构上具有特殊的异质性和动态性,无法用单一结构表示,因此其研究并未像有序蛋白质结构预测一样进展显著。而 AlphaFold 的成功却为科学家指明新的解题思路。
近日,来自剑桥大学的研究团队近期发表了一项新研究,提出了一种名为 AlphaFold-Metainference 的方法。该方法利用了 AlphaFold 预测的对齐误差 (predicted aligned error, PAE) 图与分子动力学 (molecular dynamics, MD) 模拟中的距离变化矩阵之间的相关性,从而构建无序蛋白质和含无序区域蛋白质的结构集合 (structural ensembles),为基于深度学习方法的无序蛋白质结构预测提供了新思路,同时也进一步拓宽了 AlphaFold 的适用范围。
目前,相关研究成果以「AlphaFold prediction of structural ensembles of disordered proteins」发表在国际学术期刊 Nature Communications 上。
研究亮点:
* 突破预测局限,实现高精度预测。研究证实了 AlphaFold 即便未在无序蛋白数据上训练,也能准确预测其残基间距离。
* 创新预测方法,构建结构集合。该方法利用了 AlphaFold 预测的距离作为结构约束,结合元推理框架和分子动力学模拟构建无序蛋白及含有无序区域蛋白的结构集合。
* 深化深度学习方法,拓展应用边界。该方法在处理高度无序和部分无序蛋白时表现出色,生成的结构集合与实验数据一致性显著优于单个 AlphaFold 结构,有效解决了无序蛋白结构预测难题。

论文地址:
https://www.nature.com/articles/s41467-025-56572-9
开源项目「awesome-ai4s」汇集了 200 余篇 AI4S 论文解读,并提供海量数据集与工具:
https://github.com/hyperai/awesome-ai4s
数据集:多源数据严谨验证
在深度学习模型的训练方面,由于无序蛋白质的结构集合体在数量和准确度方面都很低,但是可以基于有序蛋白的可用信息对无序蛋白质进行预测,因此研究人员采用了蛋白质数据库 (Protein Data Bank, PDB) 中大量的高分辨率折叠蛋白质结构训练深度学习模型。
在实验数据对比方面,由于获取无序蛋白质的残基间距离的试验信息具有挑战性,同时加上数据标签本身可能会影响构象集合的性质。为此研究人员采用了小角 X 射线散射 (small-angle X-ray scattering, SAXS) 数据和核磁共振 (nuclear magnetic resonance, NMR) 扩散测量,为研究提供了无标签的无序蛋白残基间距离分布信息,用于对比和验证预测结果。
另外,在进一步的验证中,研究人员还分析了通过全原子分子动力学 (all-atom molecular dynamics) 模拟获得的结构集合数据 Aβ 和 α-synuclein,并且使用 CALVADOS-2 (C2) 进行粗粒度模拟,从而进一步验证了 AlphaFold 预测距离的准确性。
模型架构:创新融合元推理方法
本研究所阐述的 AlphaFold-Metainference 方法,是用于生成代表无序蛋白质和含无序区域蛋白质天然状态的结构集合。
该方法的核心基于一项观察,即 AlphaFold 预测的残基间距离即使对于无序蛋白质也是相对准确的,因此可以在元推理框架内的分子动力学模拟中用作结构约束。简单来说,为了生成结构集合,AlphaFold-Metainference 使用了预测的距离作为分子动力学模拟中的结构约束,将 AlphaFold 距离图 (distograms) 转换为结构集合。
首先是 AlphaFold 预测距离。研究人员借助 AlphaFold 的距离图预测残基间平均距离,并通过特定公式计算预测距离和标准偏差。然后,基于 MMseqs2 进行多序列对比,使用默认设置的 AlphaFold 1.1.1 模型进行预测,期间不使用结构模板。 AlphaFold 输出残基间距离分布到 64 个等宽的 bin 中,范围从 2.15625 到 21.84375 Å,最后一个 bin 还包括了超过 21.84375 Å 的距离。
然后是结合元推理 (Metainference) 方法,所谓元推理就是一种贝叶斯推理方法,它能够根据最大熵原理,通过结合先验信息和实验数据来确定结构集合。在该阶段,研究人员将 AlphaFold 预测的距离图作为伪实验数据,运用贝叶斯元推理方法,将结构异质性和系统误差分开,如力场或前向模型不准确、数据中的随机误差以及由于系综样本量有限产生的误差等,从而确定结构集合。
在分子动力学模拟中,依据元推理能量函数进行计算,通过多副本模拟和吉布斯采样确定误差参数,最后再使用 CALVADOS-2 力场实施粗粒化模拟,实现 AlphaFold-Metainference 。
最后是距离约束选择,该阶段依据距离概率和预测对齐误差筛选 AlphaFold 预测的距离,结合蛋白质亲水性和预测局部距离差测试 (predicted local distance difference test, pLDDT) 分数确定选择标准。值得注意的是,实验利用 pLDDT 分数选取结构化区域的残基距离并非排除其作为距离约束,以优化结构集合的生成。
所有分子动力学模拟都从 AlphaFold 预测的结构开始,在 NVT 系综下进行,每个模拟设置 6 个副本,每个副本运行 100 万步,从能量最小化步骤得到的不同初始位置开始模拟。模拟采用 Langevin 积分器,时间步长为 5 fs,摩擦系数为 0.01 ps⁻¹,使用基于 Cα 的模型及 CALVADOS-2 参数和函数形式。
其中,针对高度无序和部分无序蛋白质,使用了 PULCHRA 将粗粒化集合中的所有结构转换为全原子表示,再利用 GROMACS 进行能量最小化,以获得更精确的结构。
总而言之,研究人员所展示的结果阐明了如何使用最初为预测折叠蛋白质天然状态而开发的深度学习方法来生成代表无序蛋白质天然状态的结构集合。该方法大大扩展了基于深度学习的蛋白质结构预测范围,为无序蛋白质结构预测提供了一个新思路。
实验结果:全面验证其合理性
在 AlphaFold 预测准确性方面
研究人员对比了一组 11 种既有 SAXS 测量数据又有 NMR 扩散测量数据的蛋白质,AlphaFold 预测的距离分布与 SAXS 衍生的距离分布之间具有良好的一致性。并且研究人员还添加了一种折叠蛋白质作为对照,如下图所示。
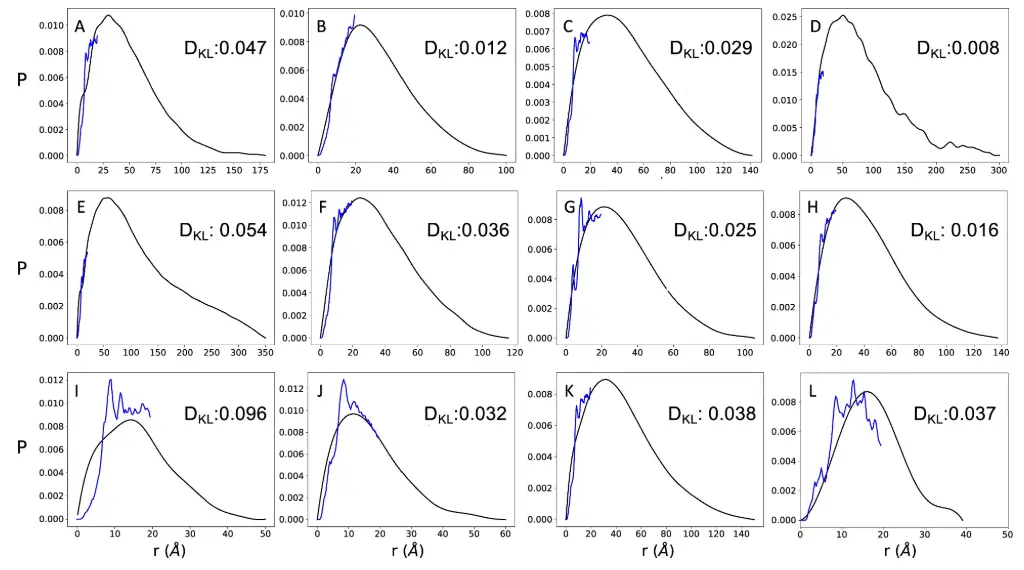
值得一提的是,由于 AlphaFold 预测的距离最远可达约 22 Å,因此 AlphaFold 预测的距离分布并未覆盖整个 SAXA 衍生的分布。结果显示,添加的对照组的 DKL 值为 0.037,与 11 种高度无序蛋白质的 DKL 值(DKL 范围为 0.008-0.096)相当,这进一步表明了 AlphaFold 对于无序和有序蛋白质的残基间距离的预测具有相当的准确性。
另外,AlphaFold 预测的距离与从 Aβ 和 α-synuclein 的 MD 集合以及从 CALVADOS-2 集合中反算出来的距离同样具有良好的一致性。
在高度无序结构集合验证方面
利用小角 X 射线散射测量可以计算出成对距离分布。研究人员比较了实验所得的距离分布与从 AlphaFold-Metainference 模拟确定的结构集合中获得距离分布,比较对象仍为上述所提到的 11 种高度无序蛋白质。
同时为了进一步比较,研究人员还展示了使用 CALVADOS-2 获得的距离分布,以及直接从单个 AlphaFold 结构中生成的 AlphaFold 派生距离分布。为了提供定量比较,研究人员发现与单个 AlphaFold 派生结构相比,AlphaFold-Metainference 与 CALVADOS-2 提供的结构集合与 SAXS 数据更为一致。
研究人员进一步使用 NMR 化学位移来比较结构集合,这些化学位移通过 CamShift 在每个时间步长进行反算获得。结果显示,部分情况下 AlphaFold-Metainference 的预测更加准确。如下图所示。
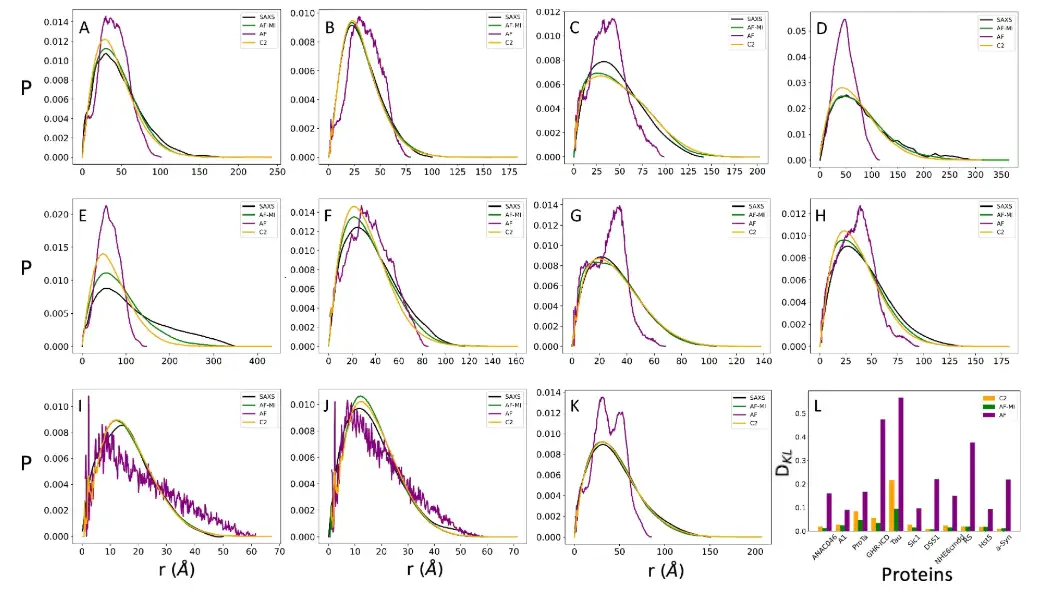
* SAXS 获得的实验成对距离的分布表示为黑线
* AlphaFold 单一结构预测表示为紫线
* AlphaFold-Metainference 结构集合预测表示为绿线
* CALVADOS-2 获得的成对距离分布表示为橙线
在部分无序结构集合验证方面
研究人员准备了一组包括 6 个既有有序结构域又有无序结构域的蛋白质,这些蛋白质具有不同的序列长度,且可以获得 SAXS 数据进行验证。
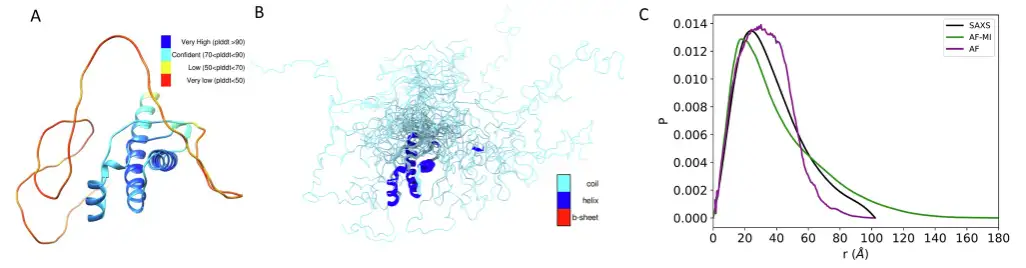
首先是 TDP-43,这是一种多功能 RNA 结合蛋白,具有模块化结构,可参与多种细胞过程,包括转录、前 mRNA 剪接和 mRNA 稳定性调节,其与肌萎缩侧索硬化症及其他神经退行性疾病有关。
实验结果发现,当应用研究人员过滤标准来选择 AlphaFold 预测的距离,并随后应用带有这些距离约束的 AlphaFold-Metainference 时,所获得的结构集合与 SAXS 数据一致性显著更好,DKL 值仅为 0.018,优于直接使用 AlphaFold 预测的结构与 SAXS 数据时 0.582 的 DKL 值。如下图所示。
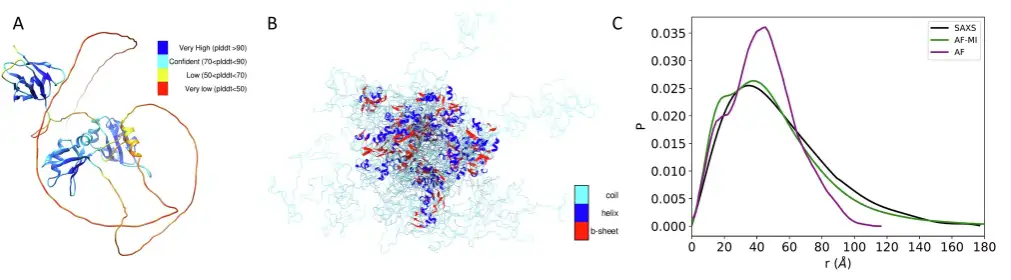
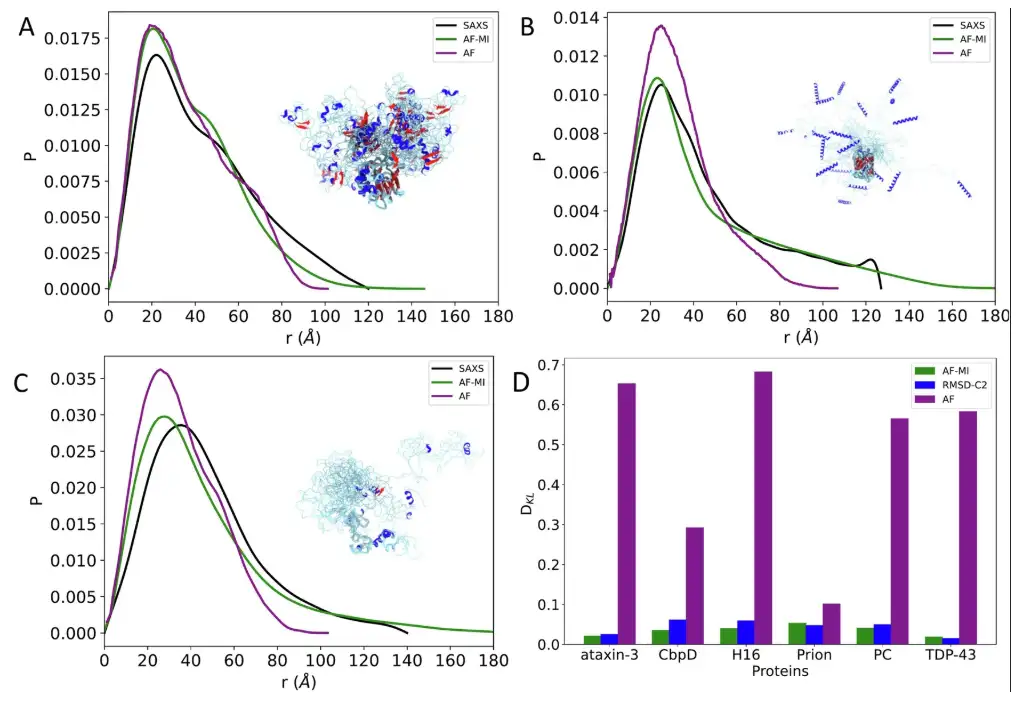
随后研究人员继续分析了 ataxin-3 和人类朊蛋白。对于前者,取得了与上述 TDP-43 相似的结果,直接从 AlphaFold 蛋白质结构数据库中使用 AlphaFold 得到的预测结构与 SAXS 数据的一致性较差,DKL 值为 0.653,而当应用过滤标准来选择 AlphaFold-Metainference 模拟的 AlphaFold 预测距离时,获得了与 SAXS 数据一致性更好的结构集合,DKL 值仅为 0.020 。如下图所示。

对于后者,直接从 AlphaFold 蛋白质结构数据库中使用 AlphaFold 得到的预测结构与 SAXS 数据的一致性较差,DKL 值为 0.1,而当应用过滤标准时,则获得了与 SAXS 数据一致性更好的结构集合,DKL 值仅为 0.053 。如下图所示。
另外,研究人员还针对其他 3 种蛋白质 CbpD 、 H16 和 PC 进行了研究,结果表明,在所有情况下,实验和反向计算的残基间距离分布之间的一致性非常好,并且相对于直接从 AlphaFold 蛋白质结构数据库中获取的 AlphaFold 单个结构有很大的改善,如下图 D 所示。
最后,在与 CALVADOS-2 方法的比较中,AlphaFold-Metainference 在 6 种蛋白质的其中 4 个(ataxin-3 、 CbpD 、 H16 和 PC),性能都更优秀,其余 2 种(TDP-43 和人类朊病毒蛋白)产生了相当的结构集合。如下图所示。
基于深度学习的无序蛋白预测进展
在过去的几年里,AlphaFold 主要被应用于预测折叠蛋白质的静态结构,也让其多为科研界所诟病,而本次研究无疑证实了其在无序蛋白质结构预测方面中同样具有潜在应用优势,同时也为无序蛋白质结构预测提供了新的研究方向。
事实上,随着 AI 与生命科学的紧密融合,针对于无序蛋白质结构预测的探讨早已是层出不穷,利用 AI 揭示生命之谜也成了现代生命科学领域中主流的手段。
比如此前曾发布在 Current Opinion in Structural Biology 上的一篇文章探讨了基于深度学习在内在无序蛋白 (Intrinsically disordered proteins , IDPs) 研究中的应用进展,并且阐述了其在无序蛋白预测、构象集合表征等方面的推动作用。
相关研究以「Deep learning for intrinsically disordered proteins:From improved predictions to deciphering conformational ensembles」为题发表。
* 论文地址:
https://www.sciencedirect.com/science/article/pii/S0959440X24001775
无独有偶,来自丹麦哥本哈根大学的研究团队以「Conformational ensembles of the human intrinsically disordered proteome」为题,在 Nature 上发布了一篇关于无序蛋白质研究的文章,其中讨论了利用多种深度学习方法预测 IDP 的无序区域、构象集合及相关属性,包括的深度学习方法如上文提到的 AlphaFold,以及蛋白质语言模型、生成对抗网络等。
*论文地址:
https://www.nature.com/articles/s41586-023-07004-5
毫无疑问,AI 的快速发展正在加速我们去了解生命的真谛。曾经英国科学家约翰·肯德鲁用 X 射线晶体学探索第一个蛋白质结构,花费了整整 12 年时间,如今 AlphaFold 只需要短短几年就能破解数以亿计个蛋白质的折叠之谜,而未来,谁又能断言我们不能掌握无序蛋白质结构预测呢?








