Command Palette
Search for a command to run...
AI for Genomics 丨空间转录组数据表征算法 SPACE,基因组学的人工智能应用

在「Meet AI4S」系列直播第二期中,我们有幸邀请到了清华大学生命科学学院张强锋实验室博士后李雨哲,他所在的张强锋实验室属于清华大学生命科学学院,同时也是清华-北大生命科学联合中心、北京结构生物学高精尖创新中心的重要组成部分。实验室的研究侧重于生命科学和人工智能算法交叉领域、 RNA 结构组技术及算法开发、单细胞的基因组测序技术及算法开发、基于冷冻电镜数据的蛋白质结构建模、以及相关人工智能算法的开发等。
本次分享,李雨哲博士以「探索基因组学的 AI 应用:以空间转录组数据表征算法 SPACE 为例」为题,分享了团队的最新研究成果,为大家介绍空间转录组学和单细胞组学研究中的 AI 方法。
HyperAI 超神经在不违原意的前提下,对李雨哲博士的本次深度分享进行了整理汇总。
点击查看完整直播回放:
AI for Science 为科学领域的研究范式带来巨大变化
今天,我分享的主题是 AI for Science,我认为 AI for Science 为整个科学领域的研究范式带来了较大的变化,接下来我将以蛋白结构的研究为例进行详细讲解。
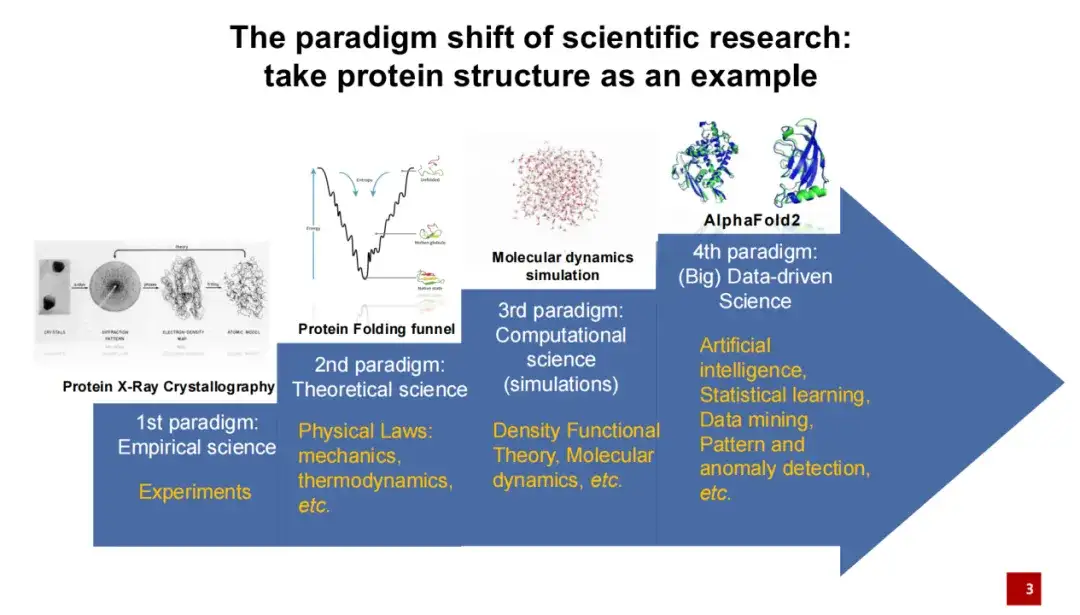
第一代蛋白结构研究范式主要通过实验手段进行,即用 X 光拍摄蛋白形成的结晶,然后再进行结构建模。
第二代蛋白结构研究范式主要是一些物理学家,将理论知识加入到蛋白结构的研究中,如蛋白如果折叠的时候能量较低,那么这种折叠相对比较稳定。
第三代蛋白结构研究范式指 20 世纪 90 年代,随着计算机技术的发展,计算机模拟逐渐应用于蛋白质结构研究,尤其是分子动力学模拟在近年来得到了广泛应用。这些模拟方法在一定程度上帮助我们更好地计算和预测蛋白质结构。最近几年,特别是在 2020 年,人工智能算法进入蛋白结构领域之后,又带来了一个突破性的进展。在 2020 年蛋白质结构预测的比赛中,相较于其他的竞赛方法,AlphaFold 2 遥遥领先。
人工智能的引入,为生命科学及整个科学研究领域带来了巨大的范式转变。与传统研究方法相比,人工智能更强调从数据出发,开展以数据驱动的科学研究。这意味着我们不再需要预先提出一个科学假设,而是直接从数据中学习和揭示自然规律。
AI for Genomics 的发展历程
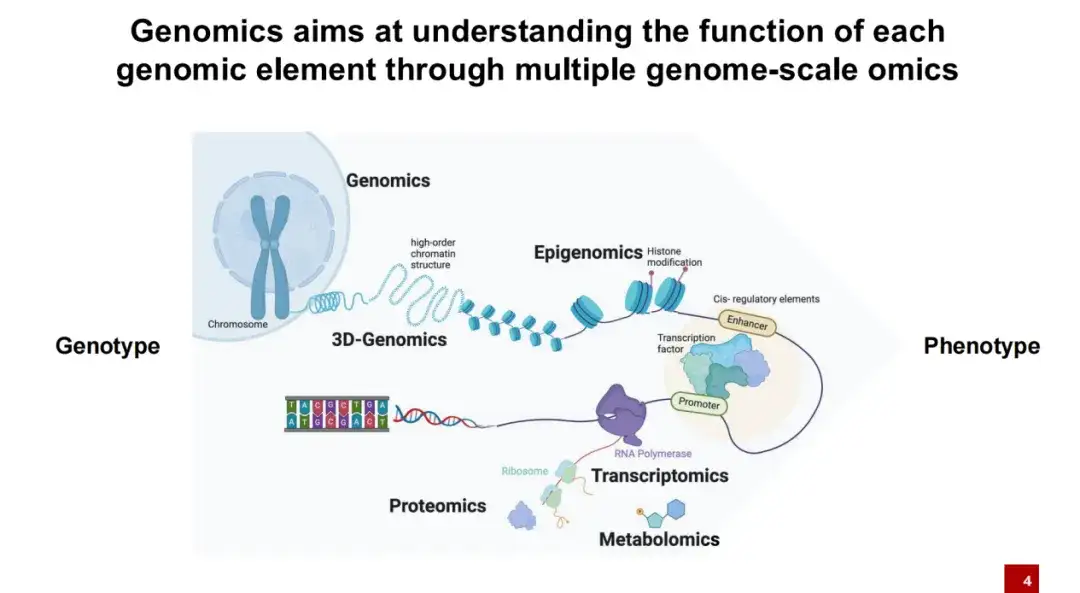
接下来的分享主要聚焦于 AI 在基因组学领域的应用。简而言之,基因组学研究主要探讨基因型 (Genotype,即体内所有的 DNA) 与表型 (Phenotype ,如身高、体重等个体特征) 之间的关系。
众所周知,DNA 在细胞内并不是裸露存在的,而是缠绕在核小体上。核小体上附着了许多组蛋白修饰,通常情况下,这些 DNA 紧密缠绕在一起,只有在特定条件下,DNA 才会暴露出来,形成开放区间。此时,转录因子等蛋白质才能结合到这些裸露的 DNA 区间上。
在之后的转录过程中,通过 RNA 聚合酶 (RNA Polymerase) 可以转录出 RNA,随后再通过核糖体 (Ribosome) 将 RNA 翻译成蛋白质,蛋白质最终在生命活动中发挥功能。基因组学的研究目标是理解各种 DNA 元件如何影响生命活动。
我们总结了自 20 世纪 50 年代 DNA 双螺旋结构被破解以来,到近期 AI for Science 发展历程中的重要事件与发展过程。其开端可以追溯到 20 世纪 50 年代 DNA 双螺旋结构的发现,以及 70 年代 Sanger 测序技术的发展。
如下图所示,蓝色部分表示各种测序技术及实验技术的发展;绿色部分表示人工智能领域的重要方法;黄色部分表示一些重要的大型研究计划及数据库的建立;红色部分则表示 AI for Genomics 领域具有代表性的方法和应用。
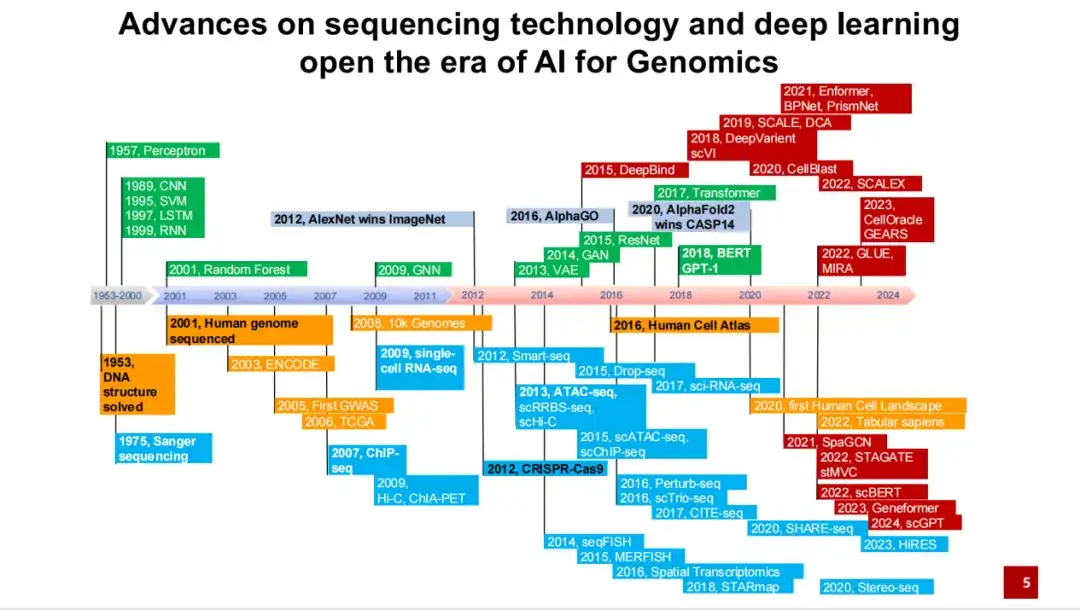
可以看到,2001 年,人类基因组计划的草图初步完成,测序了一个白人男性的全部 DNA 序列。 2012 年,AlexNet 在图像分类任务上的表现首次超越了人类,开启了人工智能领域十几年来的爆发性发展。 2016 年,提出了人类细胞图谱计划,研究逐渐从单个个体的 DNA 序列转向所有细胞。同年,基于强化学习方法的 AlphaGo 在围棋比赛中击败了人类。
对于 AI for Genomics 或 AI for Science 领域而言,一个重要的突破是 2020 年 AlphaFold 2 在 CASP 14 中取得了遥遥领先的第一名,这使得越来越多的人工智能方法开始应用于基因组学领域。
其中,Single-cell Genomics 是近年来基因组学领域的重大突破。传统的基因组学研究通常是进行整体 (Bulk) 测序,假设下图中每条线代表一种细胞,不同颜色的线代表不同的细胞类型。过去的测序方式是将整块组织进行混合测序,难以确定每条 DNA 或 RNA 来自哪个具体细胞。单细胞技术的出现使得我们不仅可以获取组织内所有的 DNA 或 RNA ,还可以明确这些 DNA 或 RNA 的具体细胞来源。由于不同细胞类型具有不同的基因表达并执行不同的功能,使我们进一步理解生命活动。
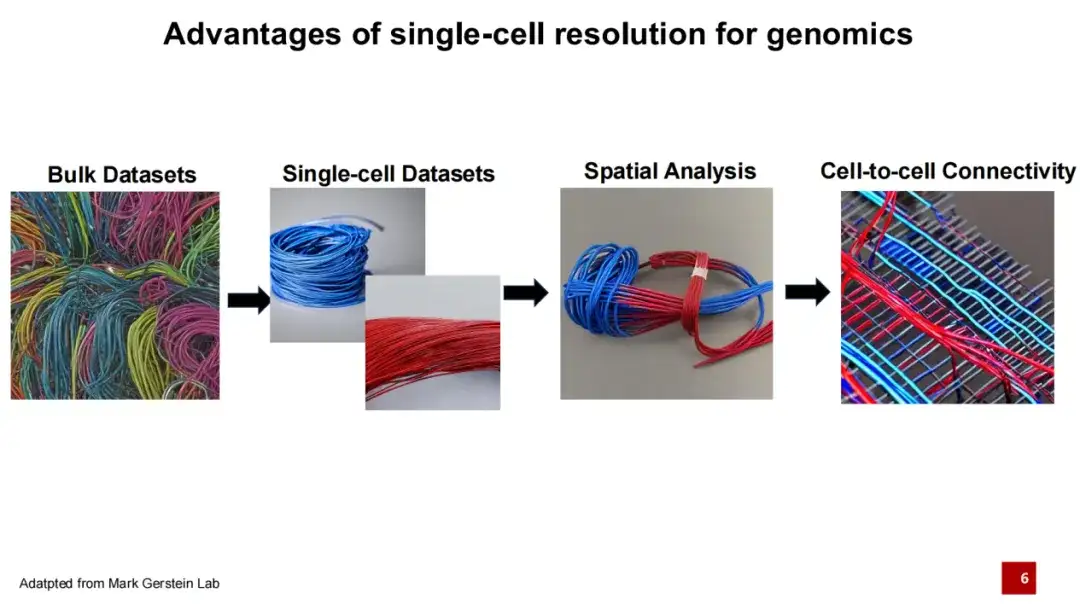
在过去 5 年中,以空间转录组为代表的空间组学技术在单细胞组学技术的基础上更进一步,不仅能够获取每个细胞类型的信息,还能确定这些细胞在空间中的分布。因为细胞间的相互作用是其功能实现的重要基础,所以进一步研究则聚焦于细胞之间如何连接。
自人类基因组计划启动以来,直到 2016 年人类细胞图谱计划的提出,其目标是完成所有人体细胞的参考图谱绘制,以帮助我们更好地理解生命活动,并为特定疾病的治疗和诊断提供支持。
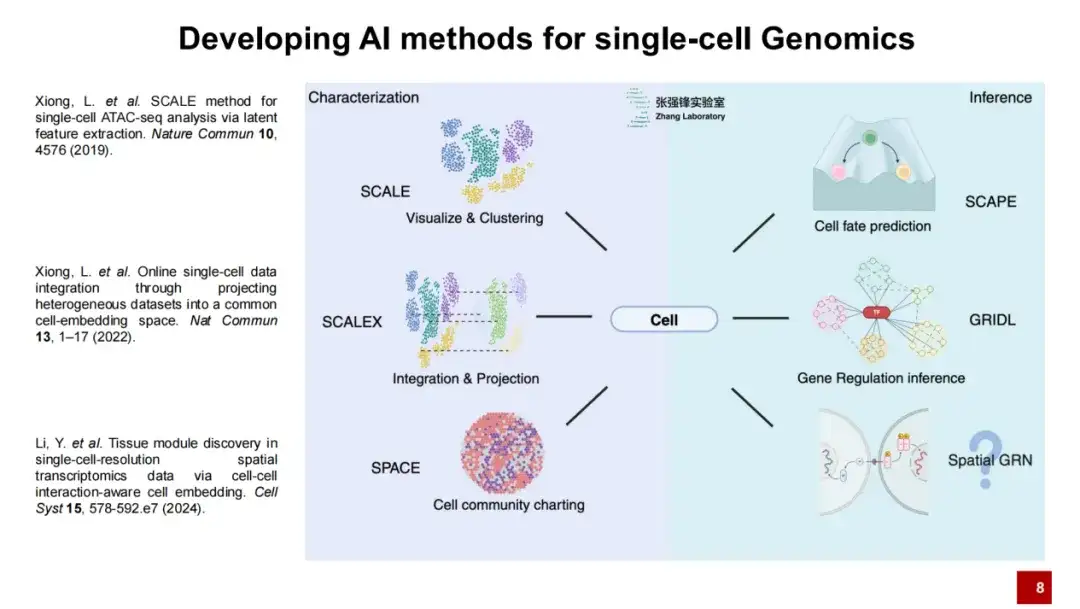
团队开发 SCALE 、 SCALEX 和 SPACE 3 种方法,进行单细胞基因组学研究
我们实验室开发了一系列人工智能方法,我们认为对 Single-cell Genomics(单细胞基因组学)来说需要两大步骤:首先是对细胞的描述,其次是对细胞的推断。
我们目前发表了 3 项工作对细胞进行描述:SCALE 、 SCALEX 和 SPACE ,其中,SCALE 主要是可视化和聚类, SCALEX 是对数据的整合和投射,SPACE 是对整个空间转录组数据组织微环境的描述。今天我主要介绍的是 SCALEX 和 SPACE 两个方法。
消除批次效应的 SCALEX 方法
SCALEX 方法是为了消除批次效应 (Batch effects),这是在基因组学研究中一个非常重要的问题。批次效应指的是,由于不同实验条件等技术因素导致不同批次实验结果存在差异。
如下图所示,即使我们分别培养了两组生物学重复的细胞,理论上对这两组细胞进行测序应得到非常相似的基因表达。然而,由于技术原因,如培养环境的差异、建库时间的不同或测序平台的差异等,最终得到的基因表达谱可能存在较大的差异,从而引入了大量技术噪声。因此,在分析数据时,需要去除这部分批次效应。

在生物学研究中,数据往往不能一次性全部收集完毕,而是通过多次实验逐步积累。因此,去除批次效应并将数据进行整合分析,以找到真正与生物学相关的因素,是基因组学或单细胞基因组学研究中的一个关键步骤。
基于此,我们开发了 SCALEX 方法,它能够将处理后的单细胞数据投射到一个广义的细胞隐空间中。 SCALEX 的框架基于变分自编码器 (VAE) 。
首先输入的是单细胞的转录组数据,然后通过不依赖批次的编码器 (batch-free encode) 将其投射到广义的细胞隐空间。
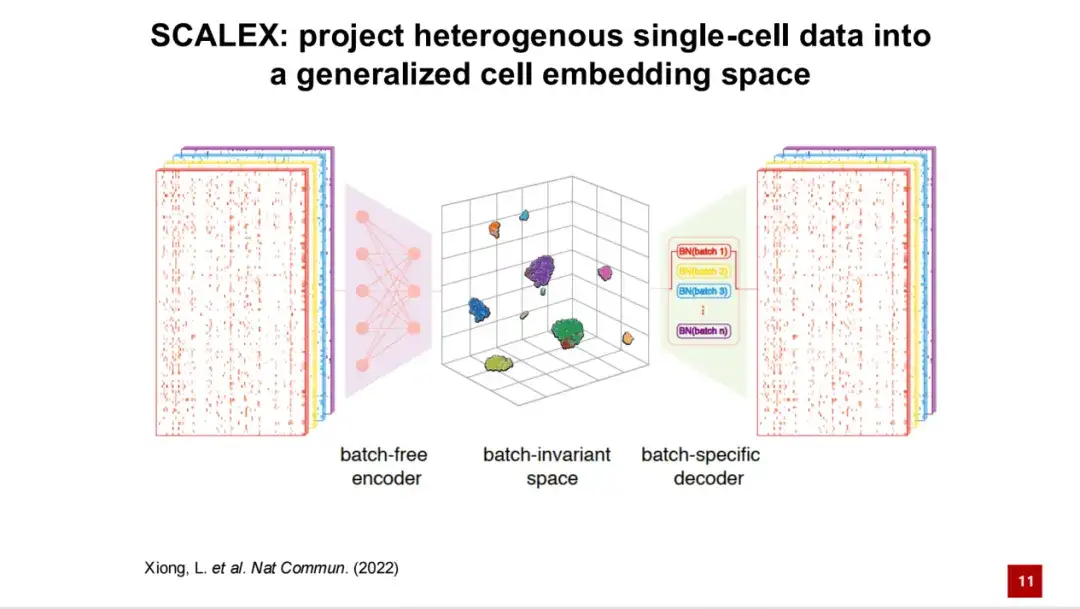
接着,再通过批次特异性的解码器 (batch-specific decoder) 将批次信息通过领域特定的批次归一化 (domain-specific batch normalization) 添加到模型中。通过这种非对称设计,生成的细胞隐空间是一个与批次无关的空间,理论上该空间不包含任何与批次相关的技术噪声。通过解码器将基因表达重新构建,并与原始输入的基因表达谱计算损失 (Loss),同时结合 KL 散度,构成 SCALEX 模型的损失函数 (Loss Function),这是一个自监督模型。
这种非对称的编码器与解码器设计有 2 个主要优点:一是得到的编码器是通用的,即新的数据可以直接通过该编码器投射到不含批次信息的细胞隐空间中,而无需重新训练模型或将新数据重新整合到已有数据中。
二是 SCALEX 更关注全局的批次效应。传统去除批次效应的方法主要是通过找到两个批次数据中的相似细胞 (cell pair),并将其配对校正,以此去除批次效应,这种方法本质上是相对局部的批次效应校正。
然而,这类方法存在一个问题,即在实际数据分析中,两个不同批次中的细胞类型可能并不完全一致,可能只有几个细胞类型是共同的,剩下的则是批次特异性的。如果强行寻找细胞配对,可能因为无法找到合适的配对细胞,进而出现过度校正 (over-correction) 的情况,将不应对齐的细胞类型强行对齐。
针对于此,我将更详细地为大家解读 SCALEX 的两大优势。
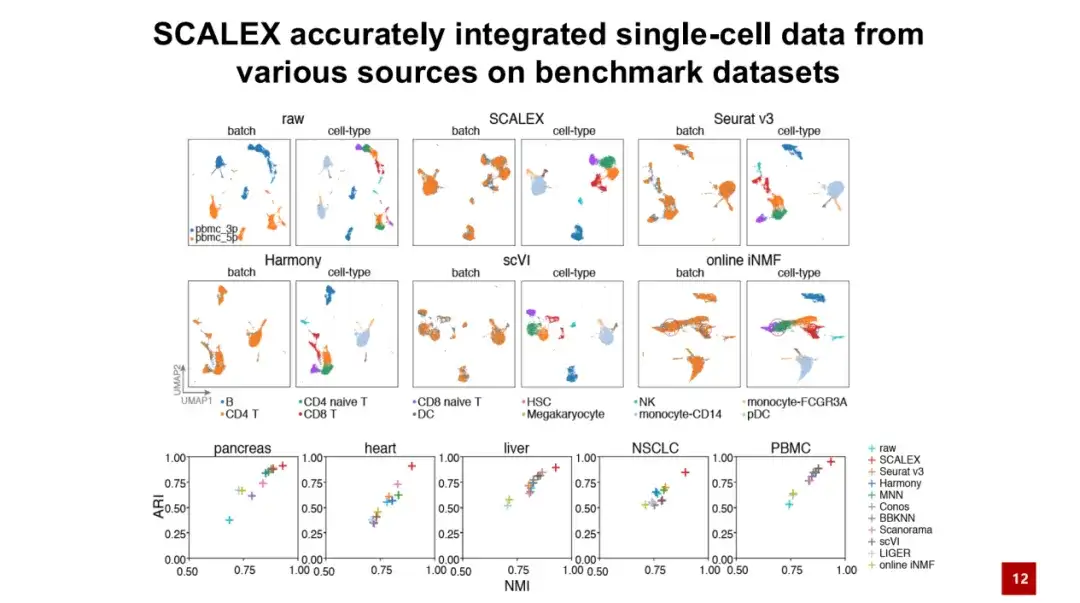
首先,我们在 5 个测试数据集上对 SCALEX 进行了基准测试 (Benchmark),结果显示 SCALEX 在精度上的表现优于现有方法。
如下图所示,batch 图表示原始且未经校正的数据,蓝色和橙色分别表示 2 个批次的数据,cell-type 表示细胞类型。可以看到,虽然这 2 个批次中有相似的细胞类型,但由于批次效应过大,原本属于同一细胞类型的细胞无法聚合在一起,导致技术因素掩盖了生物学因素,无法进行后续的生物学研究。
通过 SCALEX 整合后,这 2 个批次的细胞能够较好地聚合在一起,并且根据细胞类型进行了清晰的分离,展示了 SCALEX 在实际应用中的重要性。
SCALEX 的一个重要优势是能够处理部分细胞类型相同的 2 个批次数据,我们将这种数据称为部分重叠数据集 (partial overlapping datasets) 。如下图所示,overlap 0 表示 2 个批次中的细胞类型完全不同,overlap 4 表示 2 个批次中有 4 种共享的细胞类型。
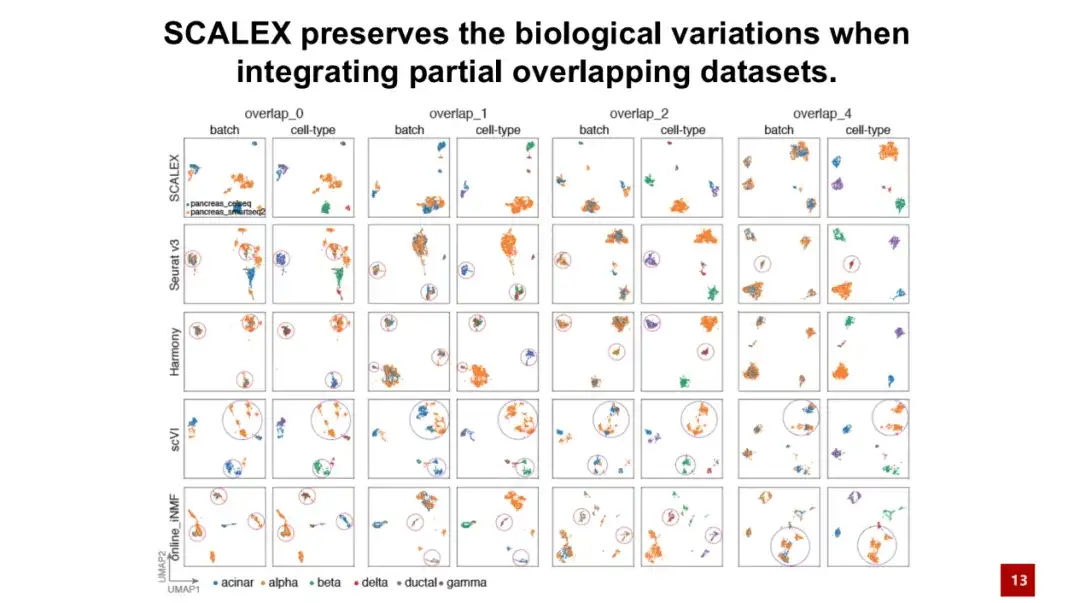
结果显示,即使在 2 个批次的细胞完全没有相同细胞类型的情况下,SCALEX 仍能很好地保持生物学差异,即 SCALEX 不会强行将不同细胞类型的细胞整合在一起,而其他类似方法可能依赖于寻找细胞配对,导致出现过度校正现象。
SCALEX 的另一个优势是通用的编码器可以直接将新数据投射到已有的无批次效应的细胞隐空间中,而无需重新训练模型。如下图所示,首先通过胰腺数据集 (pancreas datasets) 训练一个参考的细胞图谱,然后将 3 个新数据直接通过训练好的编码器投射到细胞隐空间中。图中颜色表示细胞类型,灰色的点表示已构建的参考细胞类型。可以发现,不同细胞类型在图中各自的位置得到了良好分离。
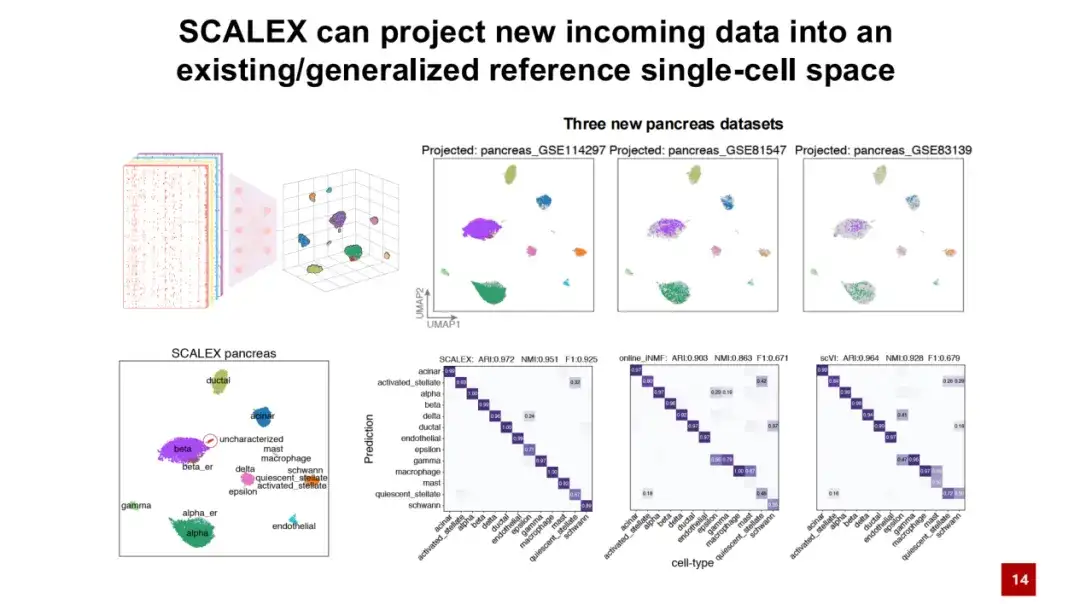
通过将特定位置周围参考细胞的标记投射到新数据细胞上,可以看到 SCALEX 在自动注释细胞类型方面表现出色。相比于目前已有的其他方法,SCALEX 展现出一个非常重要的应用,即可以直接将新数据投射到已构建的数据中,帮助我们进行数据之间的比较分析。
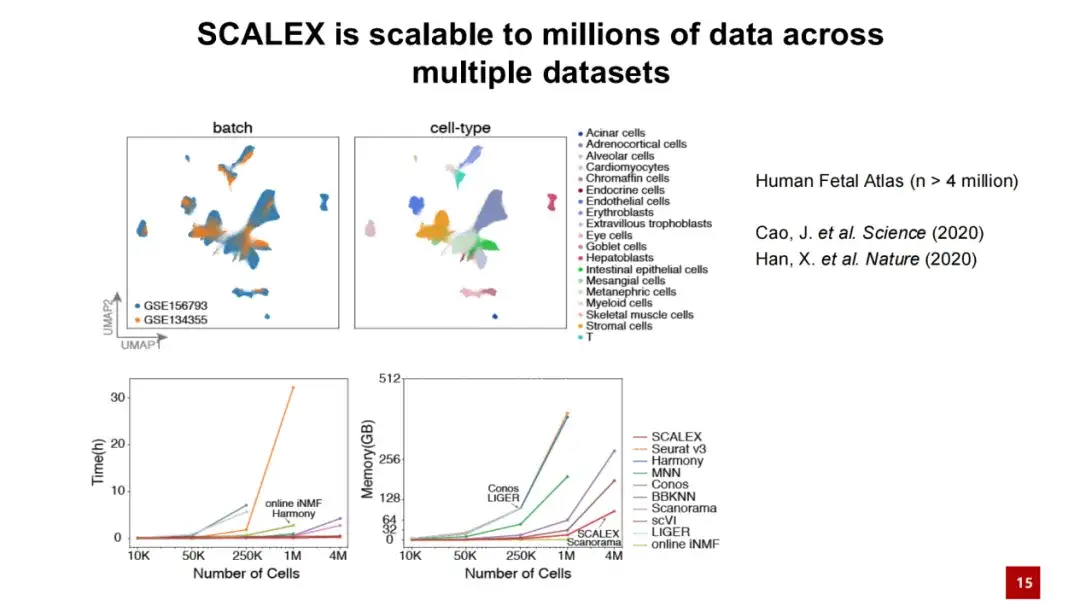
此外,SCALEX 在处理大规模数据时也表现出色。下图展示了 SCALEX 在处理 400 万细胞数据时,计算时间不超过几十分钟,内存消耗低于 100 GB 。这表明 SCALEX 具有很好的扩展性,能够用于超大规模单细胞数据的整合分析。
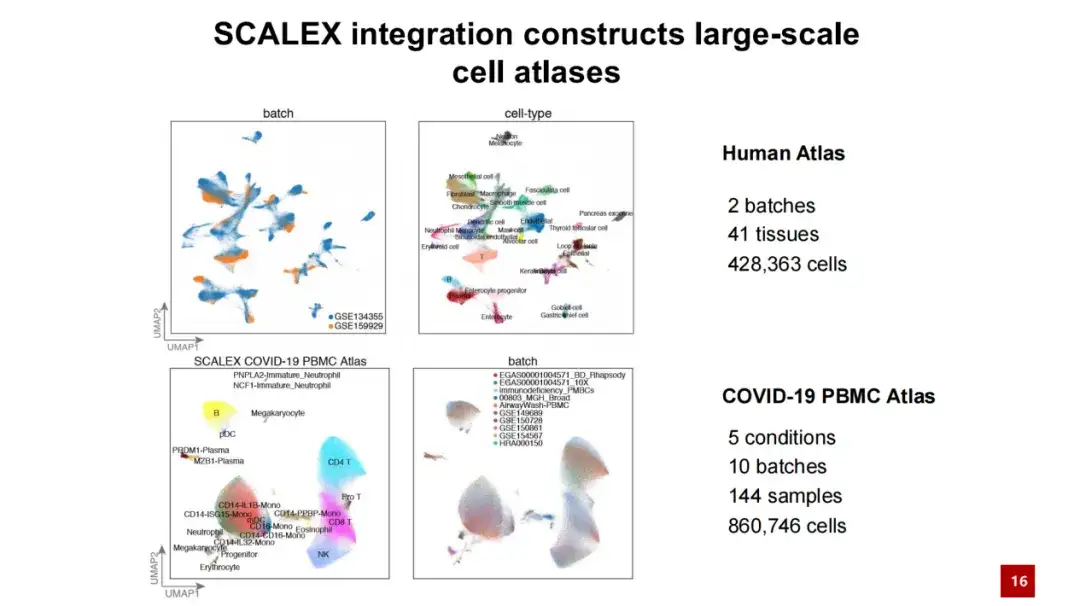
利用 SCALEX 的优势,我们构建了 2 个大规模的细胞图谱,一个是关于人类个体的细胞图谱,包含超过 40 万个细胞;另一个 是 COVID-19 PBMC 细胞图谱,包含超过 86 万个细胞和 100 多个样品。
空间转录组数据人工智能分析工具 SPACE
接下来,我将为大家介绍团队最近发表的空间转录组分析工具 SPACE 。
简单来说,空间转录组技术能够提供细胞的基因表达信息以及其在空间中的具体位置。下图展示了一个典型的空间转录组结果。在左图中,每个点代表一个细胞,颜色表示其细胞类型。这些细胞通过基因表达的降维聚类形成了 UMAP 图。在右图中,展示了每个细胞在小鼠胚胎 E16.5 数据中的实际空间位置。可以清楚地看到,细胞在空间上的分布具有很好的特异性。
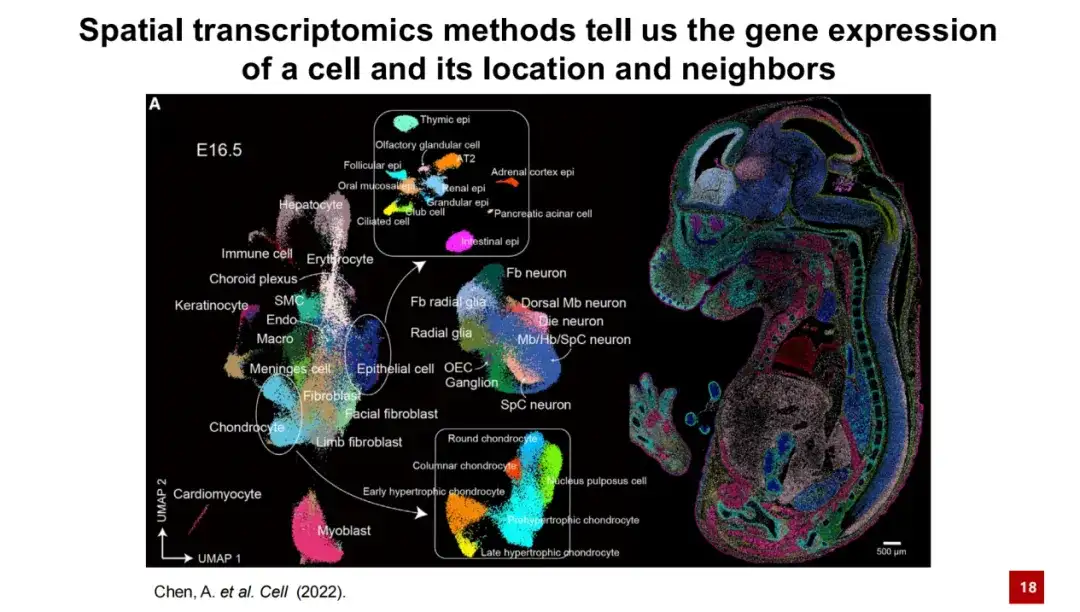
组织研究一直是生命科学研究的核心问题之一,可以说,生物学研究的长期目标之一就是理解组织的结构与其功能之间的关系。这一点很容易理解,例如大脑中不同的脑区由不同的神经元和支持细胞组成,通过复杂的细胞间相互作用执行不同功能。如有的区域负责记忆,有的区域负责学习,还有一些区域负责运动反应。
因此,空间转录组分析的一个核心问题就是识别空间中不同的细胞类型或组织模块,这一任务统称为空间聚类 (Spatial Clustering) 。
这个任务包含 2 个子任务:一是识别细胞类型,二是识别组织模块 (Tissue Module)。前者较为直观,即识别空间转录组数据中各种细胞类型,如小鼠胚胎数据中的展示;而后者则相对抽象,涉及识别组织内比组织结构更细小的区域,这些区域可能具有特定功能或由细胞组成。
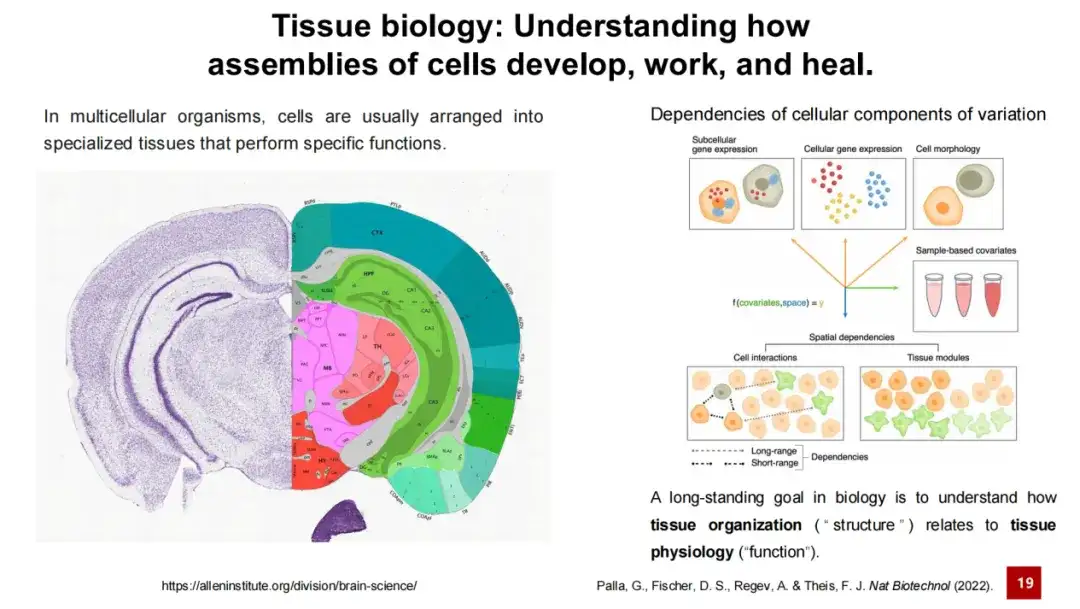
在不同研究中,研究人员会为组织模块 (Tissue Module) 赋予不同的名称,如空间域 (Spatial domain) 或细胞微环境 (Cell niche) ,其中空间域是较为常用的术语。一些研究者认为,识别组织模块就是识别具有一致空间基因表达特征的区域。
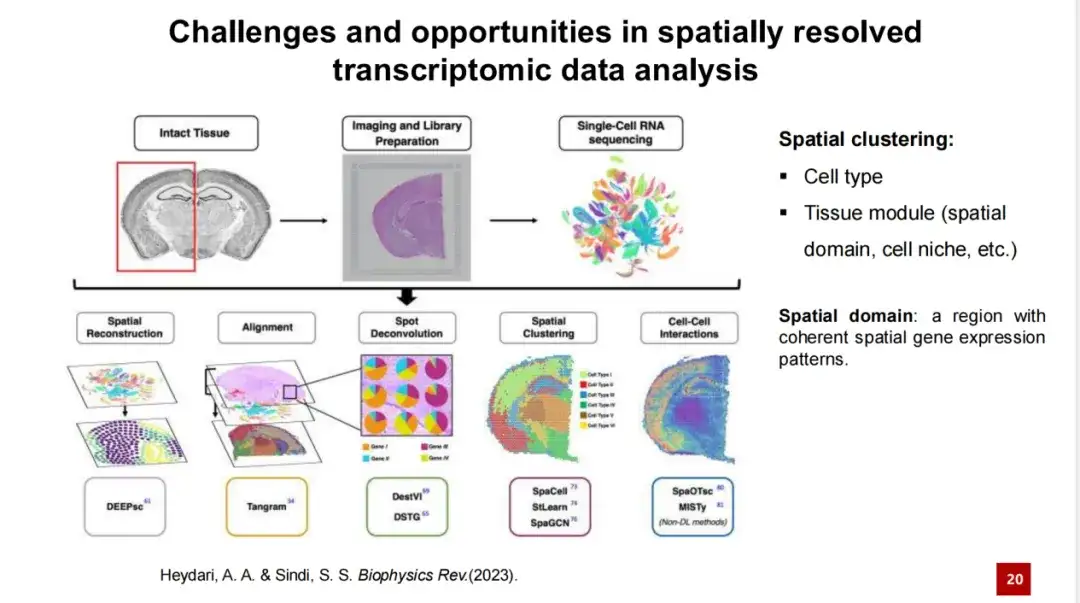
然而,这一概念存在局限性。例如,下图 A 中展示的是两个区域间存在显著的基因表达差异,但在图 B 和图 C 中,区域间的基因表达分布并不完全干净,可能存在混杂。而图 B 和图 C 展示的是空间域概念无法解决的情况。

针对于此,我们提出了 SPACE 方法,通过学习互作感知 (interaction-aware) 的细胞嵌入 (cell embeddings) 来解决空间域的问题。
SPACE 使用图自编码器 (Graph autoencoder) 框架来学习低维的细胞嵌入。
首先,我们输入空间转录组数据,并基于每个细胞的空间位置构建邻近图 (Neighboring Graph),即将每个细胞的最近邻细胞连接起来,形成一个图。在下图中,节点代表细胞,节点的特征则是细胞的基因表达特征。我们将邻近图 (Neighboring graph) 和基因表达谱 (Gene expression profiles) 输入 SPACE 的编码器,该编码器由三层 GAT 网络组成。
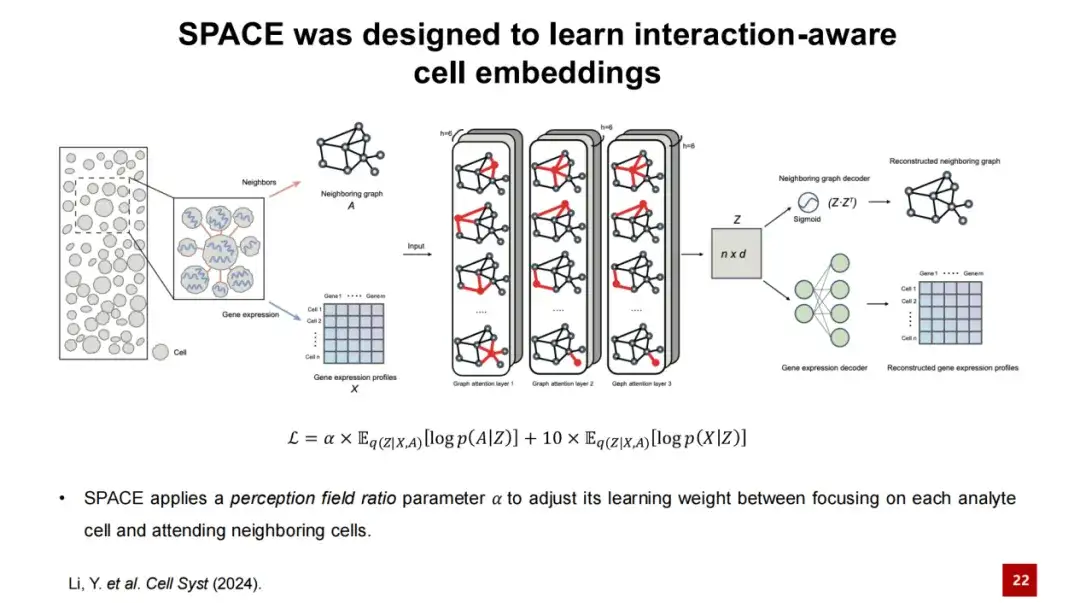
通过编码器的处理,我们可以获得每个节点的嵌入表示,并通过两个独立的解码器进行重构:一个解码器将低维细胞隐层表示重构回邻近图,另一个解码器则重构细胞的基因表达谱。 SPACE 模型的损失函数 (Loss Function) 即为这两个重构损失之和。
在此过程中,我们设计了一个感知场比例参数 α (perception field ratio paramete α),用于调整模型中两个损失函数的权重。
当 α 值较小时,模型更关注重构细胞自身的基因表达,此时获得的细胞嵌入可用于识别细胞类型;当 α 值较大时,模型更关注细胞间的相互作用,此时获得的细胞嵌入则可用于识别组织模块 (Tissue Module) 。因为低维细胞嵌入 Z 里面包含了细胞相互作用的信息,所以我们将 SPACE 得到的低维嵌入表示称为互作感知细胞嵌入 (interaction-awara cell embeddings) 。
为了识别空间细胞亚型,我们将 SPACE 应用于小鼠初级运动皮层数据集。
在下图中,左上角图展示了每个细胞在实际组织中的空间位置,其中一个点代表一个细胞,颜色表示细胞类型,这是基于基因表达生成的 UMAP 图。左下方的两个图展示了 SPACE 识别出的空间细胞亚型及其在空间中的位置。我们将这些空间细胞亚型与原始研究中提供的细胞类型进行了混淆矩阵分析(如右图所示),结果显示二者总体一致,调整后的 Rand Index (ARI) 为 0.6 。同时, SPACE 能够将星状细胞与少突胶质细胞区分得更为精细,识别出更多的细胞亚型。
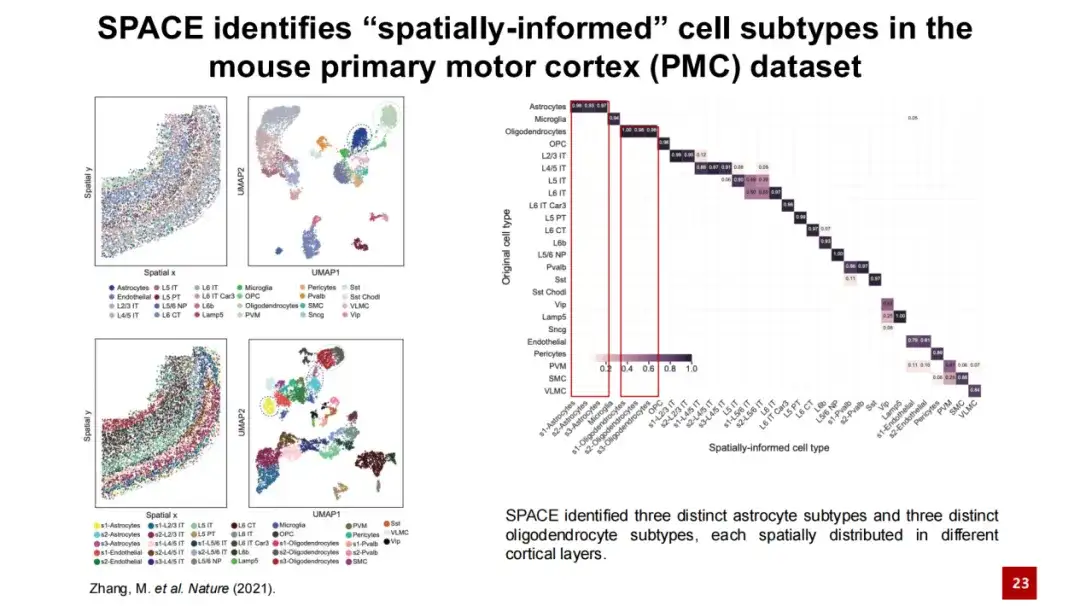
左下图展示了小鼠初级运动皮层的组织结构,Layer 表示皮层结构,WM 表示 White Matter 。可以清楚地看到从 Layer 1 到 White Matter 的分层结构。 SPACE 识别出的 3 个星状细胞亚型仅通过基因表达难以区分,在 UMAP 图中它们混合在一起。
然而,这 3 个细胞亚型在空间分布上有明确的区分:s1 细胞亚型主要分布在 Layer 1 至 Layer 4 的区域,s2 主要分布在 Layer 5 至 Layer 6 的区域,s3 则主要分布在 White Matter 中。我们统计了这 3 个星状细胞亚型周围细胞类型的比例,结果符合这一分层规律。尽管这 3 个细胞亚型在基因表达上相似,它们仍然表现出各自特异的高表达基因。

SPACE 识别出的 3 个星状细胞亚型与之前的研究高度一致。此前研究报道,星状细胞与神经元 (Neuron) 之间存在相互作用,星状细胞的分层与神经元的分层相对应。研究人员通过敲除神经元中的关键因子,发现神经元的分层结构受到破坏,而星状细胞的分层结构也相应地改变。这表明星状细胞与神经元之间存在空间特异性的相互作用及空间特异性的基因调控。
通过这一例子可以看出,SPACE 能够有效利用空间信息,准确鉴定具有空间特征的不同生物学细胞类型。
前文介绍了 SPACE 通过调整感知场比例参数 α 来改变模型的优化方向 :可以更加关注细胞自身的特征以识别细胞类型,或更加关注细胞间的相互作用信息以发现组织模块 (Tissue Module) 。
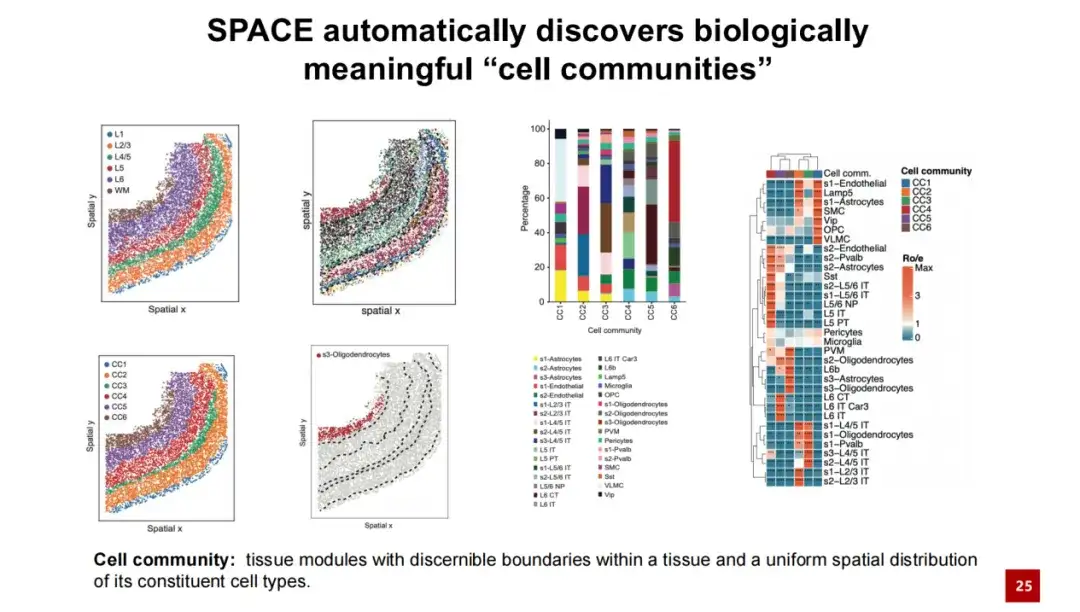
在同一数据集中,通过增大 α 值,SPACE 成功发现了组织模块 (Tissue Module),我们将其命名为细胞群落 (Cell Communities, 简称 CC) 。我们认为,SPACE 发现的组织模块具有可辨识的边界,其内部细胞类型的空间分布较为均匀一致。我们将 SPACE 发现的细胞群落与现有的组织结构进行对比,发现二者具有较好的一一对应关系。每个细胞群落内部包含不同的细胞类型,这些细胞类型在细胞群落内部的空间分布相对均匀。
我们对比了 SPACE 与现有的能够发现组织模块的方法,在 5 个数据集中进行测试分析,结果显示 SPACE 在 2 个数据集中表现优于现有最佳方法,在另外 3 个数据集上表现与最佳方法相当。我们还在常用的 Visium 人脑数据集中进行了测试分析,结果表明 SPACE 同样适用于无单细胞分辨率的空间转录组数据。
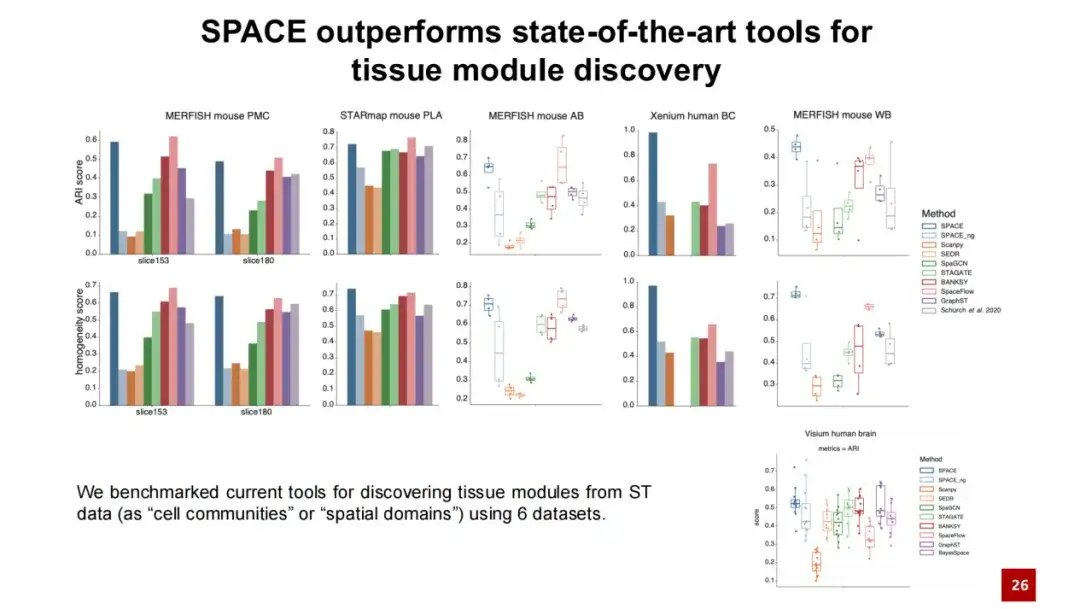
此外,我们引入了一个名为 SPACE_ng 的测试模型,ng 表示我们关闭了 SPACE 模型中的邻近图重构损失。结果显示,SPACE_ng 的表现远不如 SPACE 。
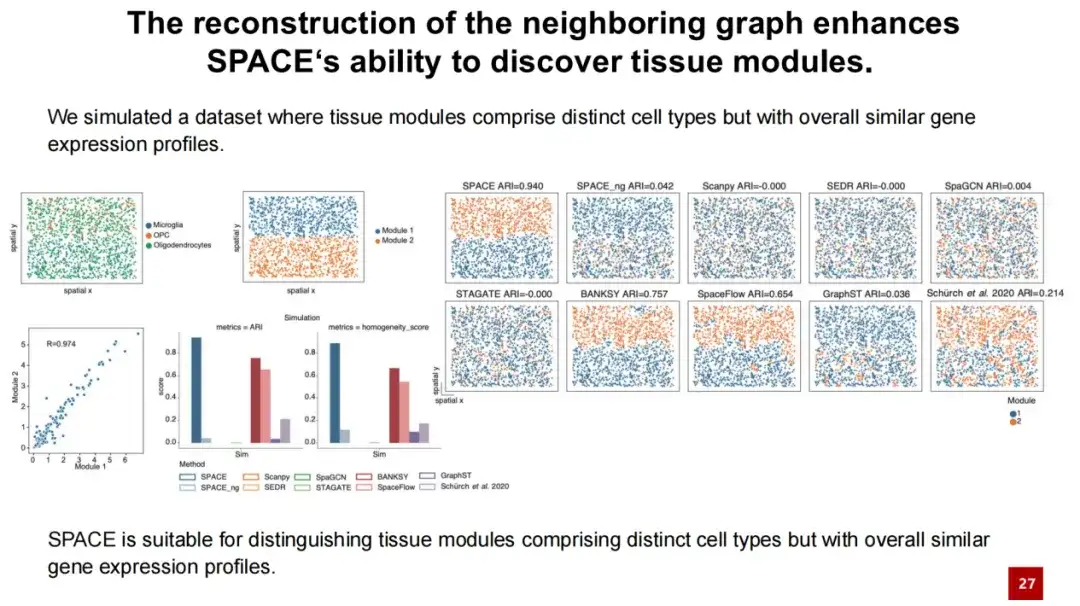
为了进一步说明 SPACE 能够很好地发现组织模块的性能来源于邻近图的重构,我们设计了一个模拟实验。我们选择了少突胶质细胞,并将小胶质细胞和 OPC 细胞均匀分布在少突胶质细胞中(见下方左上角图),形成了两个组织模块。
由于这两个组织模块中的大部分细胞都是少突胶质细胞,并且相似度极高 (collaboration = 0.97),测试结果显示,SPACE 远远优于其他方法,而 SPACE_ng 无法区分这两个组织模块。这表明 SPACE 识别组织模块的性能源于其对邻近图的重构。
我们在下游分析中观察到了类似的现象,即 SPACE 识别的细胞群落 (cell community) 的特征并非如空间域 (Spatial domain) 那样仅表现为一致的基因空间表达,而是反映了相似的邻近细胞间相互作用。
在下方热图中,每一列代表一个细胞,其颜色表示该细胞所属的细胞群落及其细胞类型。每一行代表一种细胞类型,展示了该细胞类型与其他细胞之间近邻相互作用的相对频率。从这个热图上我们可以发现,属于同一细胞群落的细胞在近邻相互作用方面表现出相似性,而这种相似性与细胞的具体类型无关。相比之下,属于不同细胞群落的细胞在近邻相互作用上则存在较大差异。
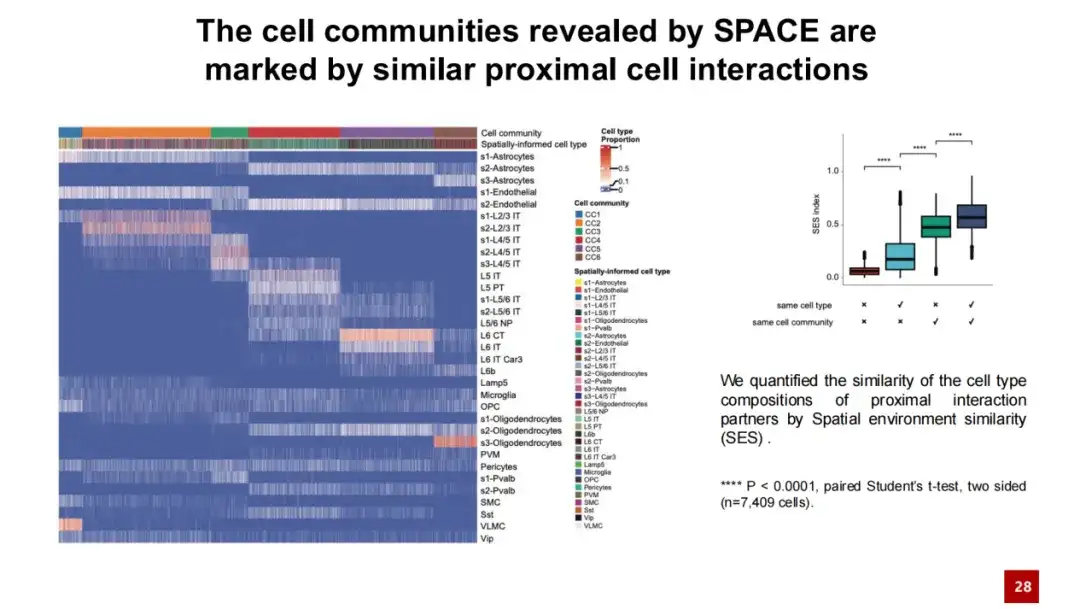
我们进一步通过余弦相似度定量计算了细胞之间相互作用的相似程度。结果表明,来源于同一细胞群落的细胞在近邻细胞间的相互作用方面具有很高的相似性,而来自不同细胞群落的细胞,其细胞外相互作用则相对不同。这些结果表明,SPACE 发现的细胞群落不仅仅是一种基因空间表达模式,更是受到近端细胞相互作用网络的影响。
我们在另外一个小鼠胎盘数据集中进行了类似分析。左侧图展示了数据集中每个细胞类型的空间位置,中间左图为人工注释的小鼠胎盘组织结构,中间右图则展示了 SPACE 发现的 5 个细胞群落。可以看到,SPACE 发现的细胞群落与人工注释的组织结构之间存在良好的一一对应关系。我们为每一个细胞群落构建了特征性的近端细胞相互作用网络,如右图所示,显示了每个细胞群落内部独特的细胞间相互作用。

以 CC1 为例,该群落主要位于母体蜕膜 (Maternal decidua) 区域。我们发现,在 CC1 中,s2 母体脱膜细胞与 s2 肝糖滋养细胞之间存在强烈的相互作用。此前的研究表明,在小鼠妊娠过程中,肝糖滋养细胞会侵入母体蜕膜区域,并与其中的母体脱膜细胞相互作用,进而引发将母体血液带入胎盘的动脉重塑过程,而这个过程对正常妊娠至关重要。
以上分析可以得出,SPACE 可以识别出生物样本中对生命过程具有重要影响的细胞间相互作用。因此,我们推测,SPACE 构建的相互作用网络可以用于优化基于配体受体的细胞通讯分析。
基于配体受体的细胞通讯分析是单细胞数据分析中的常用方法,即基于两个细胞的配体和受体的基因表达,推测它们通过配体受体对产生细胞通讯的可能性。我们首先在小鼠胎盘数据集中利用 CellChat(一种常用的细胞通讯分析方法)分析了 CC1 中细胞间的通讯。
CellChat 发现,s3 母体脱膜细胞可以通过胶原 FN1 、 THBS 等信号通路与 P-TGC 细胞类型发生细胞通讯。然而,这些信号通路都需要物理接触才能实际发生。但我们发现,这两个细胞类型在空间分布上实际上相距较远(见下方图右下角),不太可能实际发生物理接触。
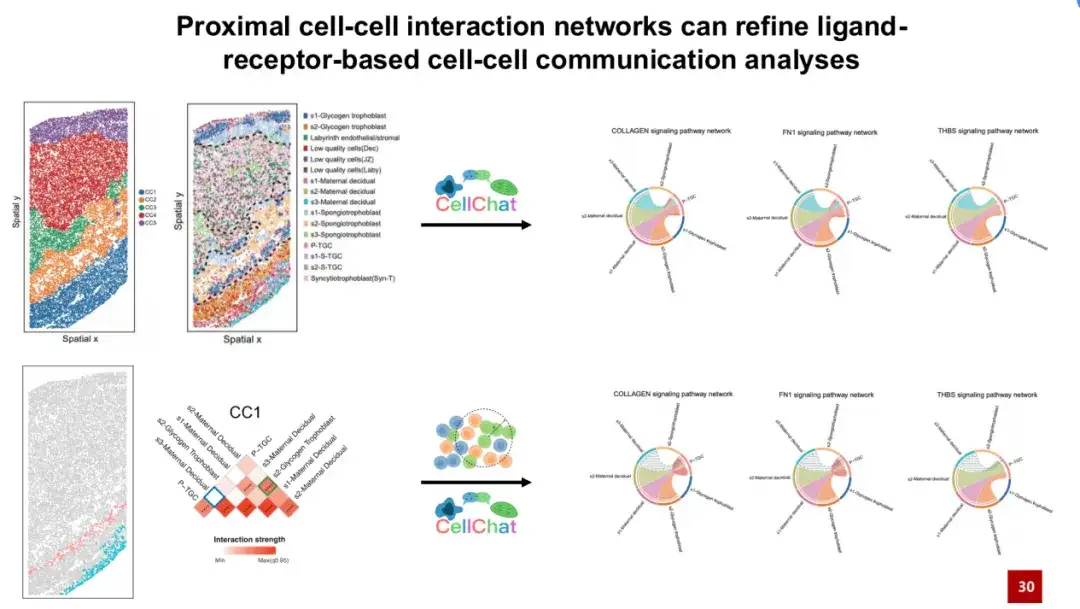
CC1 中构建的近端细胞相互作用网络也证实了这一点。蓝色框显示它们之间不太可能发生相互作用。将 SPACE 构建的特征性近端细胞相互作用网络引入 CellChat 细胞通讯分析,可以帮助我们排除空间上实际不可能发生的细胞通讯信号,从而有效减少假阳性信号。
招贤纳士
清华大学联合膜生物学国家重点实验室在杭州设立了膜结构及人工智能生物学分室。目前,该团队正在招聘从事人工智能与生物学交叉学科研究的专业人员。我们诚挚邀请有志于该领域的研究人员加入团队。若需进一步了解招聘详情,请扫描下方二维码获取更多信息。









