Command Palette
Search for a command to run...
入选 ICML!人大团队将等变图神经网络用于靶蛋白结合位点预测,性能最高提升 20%
生命系统中,几乎所有生物和药理过程都涉及受体(靶蛋白)和配体(小分子)之间的相互作用,这些相互作用发生在靶蛋白结构的特定区域,称为「结合位点」——预测靶蛋白的结合位点在药物发现等各种下游任务中起着基础性的作用。
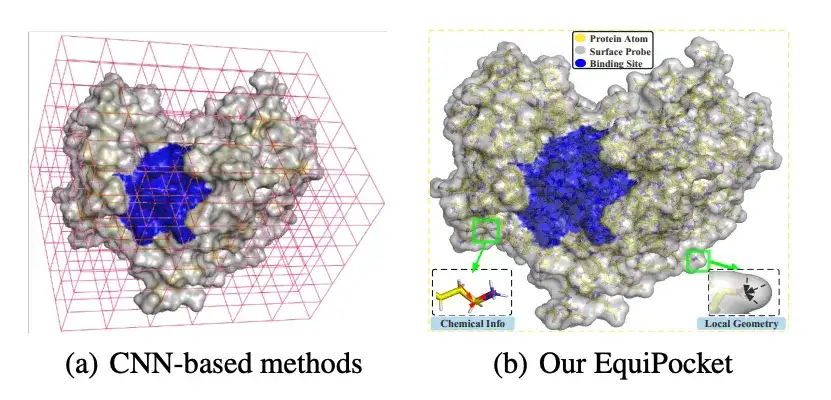
近年来,受深度学习突破的启发,卷积神经网络 (CNN) 已经成功应用于配体结合位点预测。基于 CNN 的方法通过将蛋白质的原子空间聚类到最近的体素 (voxel) 中,将蛋白质视为三维图像,然后将结合位点预测建模为 3D 网格上的目标检测问题或语义分割任务。这些方法具有一定的优越性,但仍存在挑战,例如,在表示不规则蛋白质结构方面存在缺陷;对旋转敏感;对蛋白质表面几何特征描述不足;对蛋白质大小变化不敏感。
为此,来自中国人民大学高瓴人工智能学院的研究团队,近期在 AI 领域顶级学术会议 ICML 2024 上,发表了题为「EquiPocket: an E(3)-Equivariant Geometric Graph Neural Network for Ligand Binding Site Prediction」的研究论文。该研究首次将 E(3) 等变图神经网络 (GNN) 应用于配体结合位点预测,提出名为 EquiPocket 的框架,解决了基于 CNN 的方法所遇到的挑战。
研究亮点:
* 首次将 E(3) 等变 GNN 应用于配体结合位点预测
* 与传统的基于 CNN 的方法相比,EquiPocket 不需要体素化过程,能够对不规则的蛋白质结构进行建模,对任何欧氏变换都不敏感,从而解决了「在表示不规则蛋白质结构方面存在缺陷」以及「对旋转敏感」等挑战
* 在有代表性的基准方法上进行的广泛实验,证明了 EquiPocket 相比于当前最先进方法的优越性,有助于药物发现等各种下游任务
论文地址:
https://openreview.net/forum?id=1vGN3CSxVs
开源项目「awesome-ai4s」汇集了百余篇 AI4S 论文解读,并提供海量数据集与工具:
https://github.com/hyperai/awesome-ai4s
数据集:多个专业数据集综合验证
研究人员选取了多个专业数据集,并使用每个数据集的 mlig 子集进行评估,这些子集包含用于结合位点预测的相关配体。
其中,scPDB 是用于结合位点预测的著名数据集,包含由 VolSite 生成的蛋白质、配体和三维空腔结构。本研究使用 2017 年发布的版本进行训练和交叉验证,该版本包含 17,594 个结构、 16,034 个条目、 4,782 种蛋白质和 6,326 种配体。
PDBbind 是一个研究蛋白质-配体复合物常用的数据集,包含蛋白质、配体、结合位点的三维结构以及实验室确定的准确结合亲和力结果。本研究使用 2020 年版本进行评估,该版本包括两个部分:普通集 (14,127 个复合物) 和精炼集 (5,316 个复合物) 。普通集包含所有的蛋白质-配体复合物,精炼集则从普通集中挑选出质量更好的化合物,用于实验测试。
COACH 420 和 HOLO4K 是用于结合位点预测的 2 个测试数据集,首次由 (Krivák & Hoksza, 2018) 引入。
模型架构:EquiPocket 整体框架由三大模块构成
EquiPocket 的整体框架由 3 个模块组成,如下图所示:
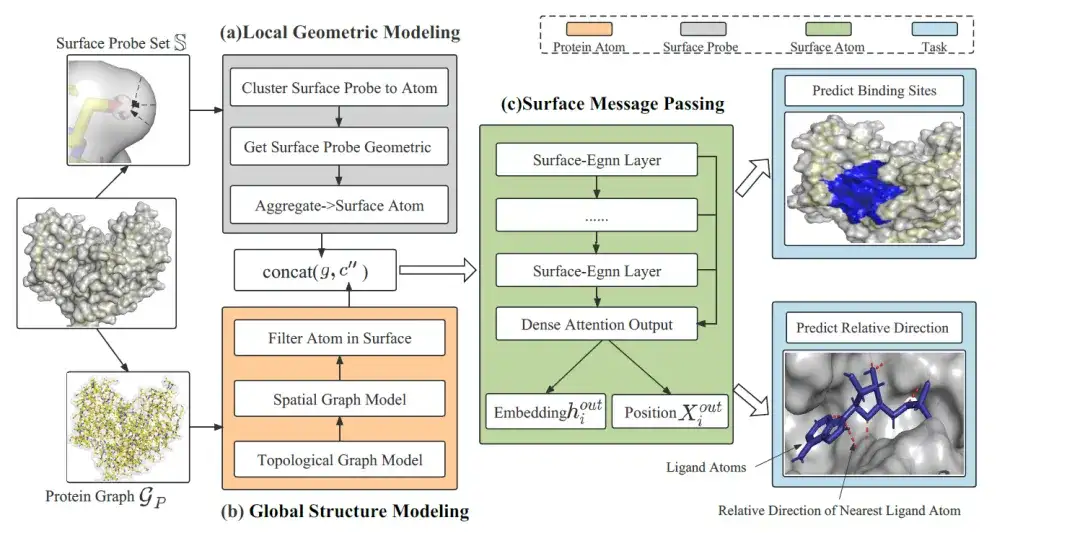
第一个模块为局部几何建模模块 (Local Geometric Modeling),用于提取每个表面原子的局部几何信息;第二个模块为全局结构建模模块 (Global Structure Modeling),用于描述蛋白质的化学和空间结构;最后一个模块为表面信息传递模块 (Surface Message Passing),通过传递表面原子上的等变信息,进而捕获表面的几何形状。
局部几何建模模块
每个蛋白质原子的局部几何形状决定了其附近区域是否适合成为结合位点的一部分。
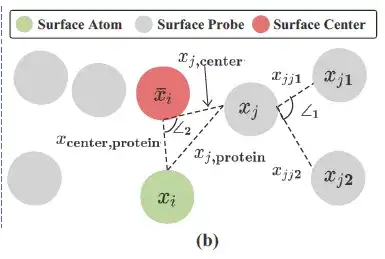
如上图所示,研究人员利用每个蛋白质表面原子 (Surface Atom,上图绿色) 周围的表面探针 (Surface Probes,上图灰色) 来描述局部几何信息。具体来说,每个表面原子 i ∈ VS,其周围的表面探针由 S 的一个子集返回,即:
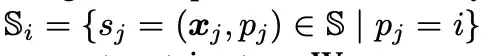
研究人员基于 Si 构建几何信息,将 Si 中所有 3D 坐标的中心/平均值 (Surface Cente,上图红色) 记为 xi¯。
全局结构建模模块
尽管结合位点主要由表面原子组成,但蛋白质的整体结构通常会影响配体的相互作用以及结合位点的形成,因此需要对其进行建模。
研究人员通过 2 个连接的过程来实现这一目标:化学图建模和空间图建模,由此形成的全局结构建模模块负责处理整个蛋白质的信息,包括原子类型、化学键、相关空间位置等。
表面信息传递模块
给定表面原子的局部几何特征以及全局编码特征,该模块将在表面图上进行等变信息传递,以更新蛋白质表面原子的全部特征。
研究结果:EquiPocket 相比基线模型性能提升 10-20%
在实验中,研究人员选择用以下基线模型与 EquiPocket 进行比较:
* 基于几何的方法:Fpocket
* 机器学习方法:P2rank
* 基于 CNN 的方法:DeepSite, Kalasanty , DeepSurf, RecurPocket
* 基于拓扑图的模型:GAT , GCN 和 GCN2
* 基于空间图的模型:SchNet, EGNN
评估模型采用的指标包括 DCC (预测的结合位点中心与真实的结合位点中心之间的距离) 、 DCA (预测的结合位点中心与任何配体网格之间的最短距离) 和失败率 (没有任何预测的结合位点中心的采样率) 。下表是在 COACH 420 、 HOLO4K 和 PDBbind 上进行结合位点预测的结果。
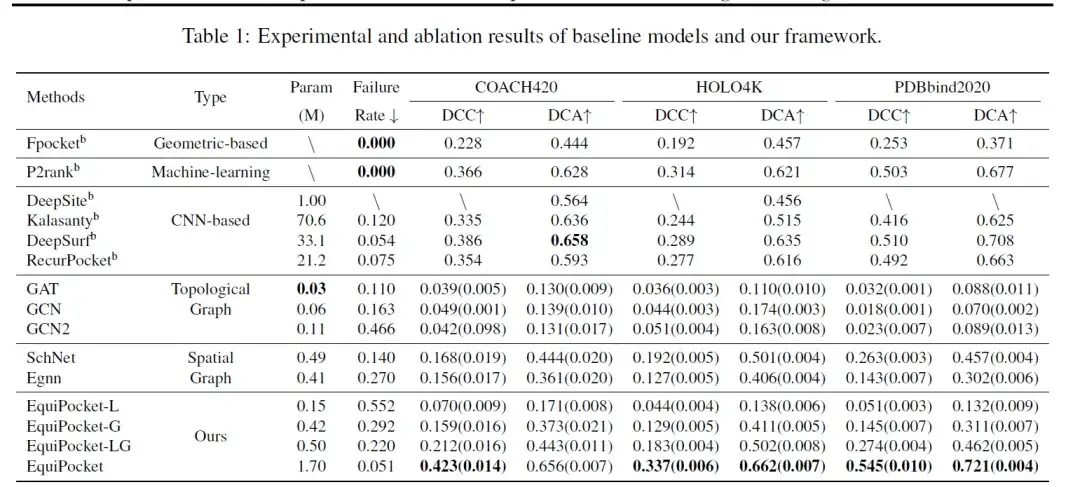
如表中数据所示,基于几何的方法 Fpocket 的性能较差,由于该方法仅使用蛋白质的几何特征,故失败率为 0;机器学习方法 P2rank 结合随机森林与蛋白质表面的几何信息,从而显著提升了性能。
基于 CNN 的方法 (DeepSite, Kalasanty , DeepSurf, RecurPocket) 性能要远高于基于几何的方法,其中 DCC 和 DCA 提升超过 50%,但需要大量的参数和计算资源。这其中,早期提出的方法 DeepSite 和 Kalasanty 受到蛋白质大小变化和处理大蛋白质能力不足的限制,可能导致预测失败。
对于图模型,拓扑图模型 (GCN 、 GAT 、 GCN2) 的性能较差,主要是因为它们只考虑原子和化学键信息,忽略了蛋白质的空间结构;空间图模型 (SchNet, EGNN) 的性能通常优于拓扑图模型。 EGNN 利用原子的性质以及它们的相对/绝对空间位置,效果更好;SchNet 仅基于原子的相对距离更新嵌入,但是空间图模型的性能比基于 CNN 和基于几何的方法差,因为前者无法获得足够的几何特征,也不能解决蛋白质大小变化的问题。
以上结果表明,蛋白质表面的几何信息和多层级结构信息对于结合位点预测至关重要。
此外,这也反映了当前 GNN 模型的局限性,即从蛋白质表面收集足够的几何信息很难或者所需的计算资源过大,难以应用于蛋白质等大分子系统。因此,EquiPocket 框架不仅能够从原子层面更新化学和空间信息,还能够有效地收集几何信息,而无需过多计算资源支持,其性能比先前的结果提高了 10-20% 。
从小分子配体到生物大分子,AI 深度解读蛋白质结构
每个植物、动物和人类细胞内都有数十亿个分子机器,由蛋白质、核酸、糖类等分子组成,其中的任何一个部分都难以单独发挥作用——只有了解它们如何在数百万种组合中相互作用,才能更加深刻地理解生命。
今年 5 月,Google DeepMind 重磅发布了 AlphaFold3 模型,该模型能够对包括蛋白质、核酸、小分子、离子和修饰残基在内的复合物进行联合结构预测。蛋白质与小分子配体的相互作用是药物作用机制的核心,AlphaFold3 通过其先进的深度学习算法,能够精确预测蛋白质与配体结合的三维结构,准确性远超现有的对接工具。
新药开发方面,通过 AlphaFold3 预测的蛋白质-配体结构,研究人员可以更有效地筛选和设计新药候选物,加速药物发现流程;现有药物优化方面,该工具还可以用于优化现有药物,通过改进其与靶蛋白的结合模式来增强疗效或减少副作用。
除了小分子配体,蛋白质发挥生物学功能也需通过与 DNA 、糖类等生物大分子的相互结合。目前,数以千计的蛋白质结构复合物已通过实验方法存放在蛋白质数据库中,然而,传统实验方法耗时且昂贵,基于机器学习的预测方法则能让挑战迎刃而解。
今年 2 月,来自南京农业大学的研究团队在生物学领域重要期刊 Briefings in Bioinformatics 在线发表了题为「ULDNA: Integrating Unsupervised Multi-Source Language Models with LSTM-Attention Network for High-Accuracy Protein-DNA Binding Site Prediction」的研究论文。针对蛋白质-DNA 结合位点预测问题,开发了一种新的深度学习预测方法 ULDNA 。
论文地址:
https://academic.oup.com/bib/article/25/2/bbae040/7606634

ULDNA 的核心思想是利用蛋白质大语言模型针对序列设计特征表示,再结合注意力机制的长短期记忆网络 (LSTM-Attention Network) 训练 DNA 结合位点预测模型。研究人员选取了 PDNA-128 、 PDNA-316 、 PDNA-335 等 7 个基准数据集 (蛋白质序列数目从 40 到 600 不等),对 ULDNA 进行了全面测试。实验结果表明,ULDNA 在所有数据集上均表现卓越,预测性能明显优于其他 9 种主流方法。
DNA 之外,糖在所有生物体的细胞表面普遍存在,它们与多种蛋白质家族如凝集素、抗体、酶和转运蛋白相互作用,调节免疫反应、细胞分化和神经发育等关键生物学过程。理解糖类与蛋白质的相互作用机制是开发糖类药物的基础。然而,糖类结构的多样性和复杂性,尤其是它们与蛋白质结合位点的多变性,给实验数据的获取和药物设计带来了挑战。
就在近日,中国科学院团队开发了一种深度学习模型 DeepGlycanSite,能够准确预测给定蛋白质结构上的糖结合位点。 DeepGlycanSite 将蛋白质的几何和进化特征融入基于 Transformer 架构的深度等变图神经网络中,其性能显著超越了之前的先进方法,并能有效预测各种糖类分子的结合位点。
结合诱变研究,DeepGlycanSite 揭示了重要 G 蛋白偶联受体的鸟苷-5’-二磷酸糖识别位点。这些发现表明 DeepGlycanSite 对于糖结合位点预测具有重要价值,并可以深入了解具有治疗重要性蛋白质的糖类调节背后的分子机制。

该研究以「Highly accurate carbohydrate-binding site prediction with DeepGlycanSite」为题,于 2024 年 6 月 17 日发布在 Nature Communications 。
论文地址:
https://www.nature.com/articles/s41467-024-49516-2
总而言之,蛋白质是生命体中的重要分子,对细胞的结构和功能发挥着关键作用,研究蛋白质的结构对于理解生命过程、揭示疾病机制以及药物研发具有重要意义。如今,机器学习为科学家理解生命的奥妙打开了一扇新的大门。
参考资料:
1.https://openreview.net/forum?id=1vGN3CSxVs
2.https://mp.weixin.qq.com/s/aGzcr0ncQA-jBy-vTGC35Q
3.https://www.jiqizhixin.com/articles/2024-05-09
4.https://news.njau.edu.cn/2024/0








