Command Palette
Search for a command to run...
神经网络替代密度泛函理论!清华研究组发布通用材料模型 DeepH,实现超精准预测

在材料设计中,了解其电子结构与性质是预测材料性能、发现新材料、优化材料性能的关键。过去,业界广泛使用密度泛函理论 (DFT) 来研究材料电子结构和性质,其实质是将电子密度作为分子(原子)基态中所有信息的载体,而不是单个电子的波函数,从而将多电子体系转化为单电子问题进行求解,既简化了计算过程,又可以确保计算精度,能更准确地反映孔径分布。
然而,DFT 的计算成本极高,通常只能用于研究小尺寸的材料系统。受到材料基因组倡议的启发,科学家们开始尝试利用 DFT 构建庞大的材料数据库,虽然目前只收集到了有限的数据集,但这已经是一个了不起的开始。以此为开端,随着 AI 技术带来的全新变革,研究人员开始思考,「将深度学习与 DFT 进行结合,让神经网络深入学习 DFT 的精髓,能否带来一场革命性突破?」
这正是深度学习密度泛函理论哈密顿量 (DeepH) 方法的核心。通过将 DFT 的复杂性封装在一个神经网络中,DeepH 不仅能够以前所未有的速度和效率进行计算,而且随着训练数据的增加,其智能也在不断提升。近日,来自清华大学物理系的徐勇、段文晖研究组成功利用其原创的 DeepH 方法,发展出 DeepH 通用材料模型,并展示了一种构建「材料大模型」的可行方案,这一突破性进展为创新材料发现提供了新机遇。
相关研究以「Universal materials model of deep-learning density functional theory Hamiltonian」为题,已发表于 Science Bulletin 。

论文地址:
https://doi.org/10.1016/j.scib.2024.06.011
开源项目「awesome-ai4s」汇集了百余篇 AI4S 论文解读,并提供海量数据集与工具:
https://github.com/hyperai/awesome-ai4s
通过 AiiDA 构建大型材料数据库,针对性排除磁性材料干扰
为了证明 DeepH 通用材料模型的普适性,该研究通过自动交互式基础设施和数据库 (AiiDA) 构建了一个包含 104 种固体材料的大型材料数据库。
为了展示多样化的元素组成,该研究还选择了元素周期表的前四行,从而排除掉了从 Sc 到 Ni 的过渡元素,以避免磁性材料干扰,并排除了稀有气体元素。候选材料结构则来源于 Materials Project 的数据库。除了基于元素类型进行过滤之外,候选材料在材料项目中被进一步细化为只包括那些标有「非磁性」的材料。为简单起见,在晶胞中包含超过 150 个原子的结构被排除在外。
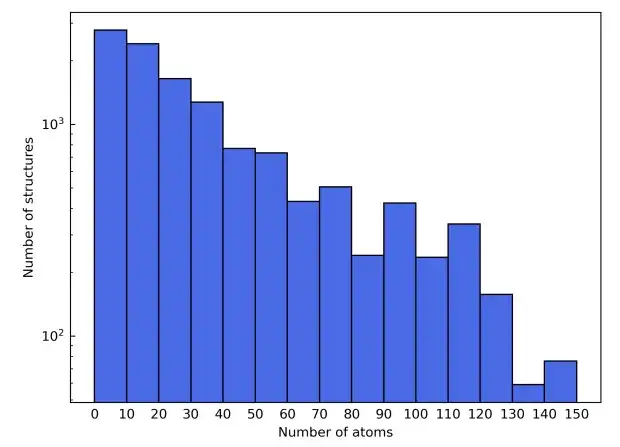
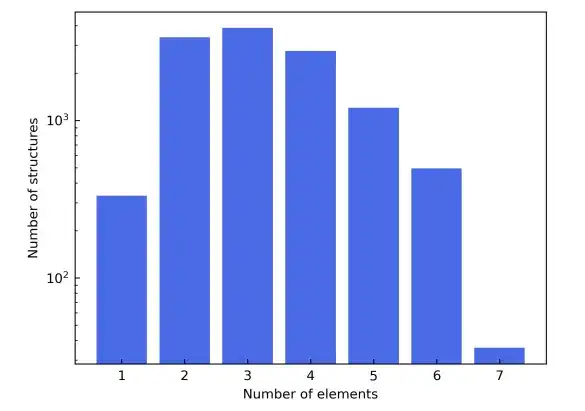
作为这些过滤标准的结果,最终的材料数据集由总共 12,062 个结构组成。在训练过程中,数据集按 6:2:2 的比例划分为训练集、验证集和测试集。接下来,该研究利用 AiiDA(自动化交互式基础设施和数据库)的框架开发了一个高通量的工作流程来进行密度泛函理论计算,并用它来构建材料数据库。
以 DFT 哈密顿量为目标,用 DeepH-2 方法训练 DeepH
研究认为,DFT 哈密顿量 (DFT Hamiltonian) 是理想的机器学习目标。
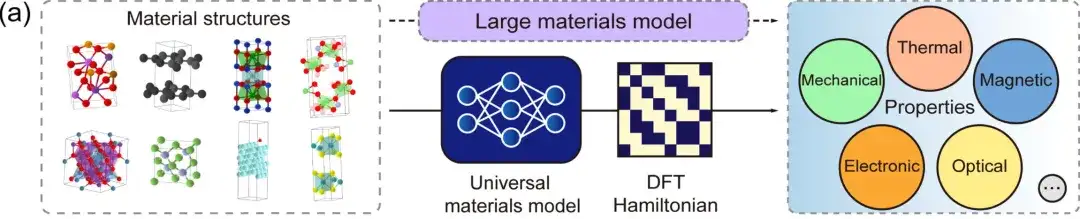
首先,DFT 哈密顿量是可以直接从总能量 (total energy) 、电荷密度 (charge density) 、能带结构 (band structure) 、物理响应 (physical responses) 等物理量中导出的基本量,DeepH 通用材料模型则可以接受任意材料结构作为输入,并生成相应的 DFT 哈密顿量,从而可以直接推导各种材料特性,如上图所示。
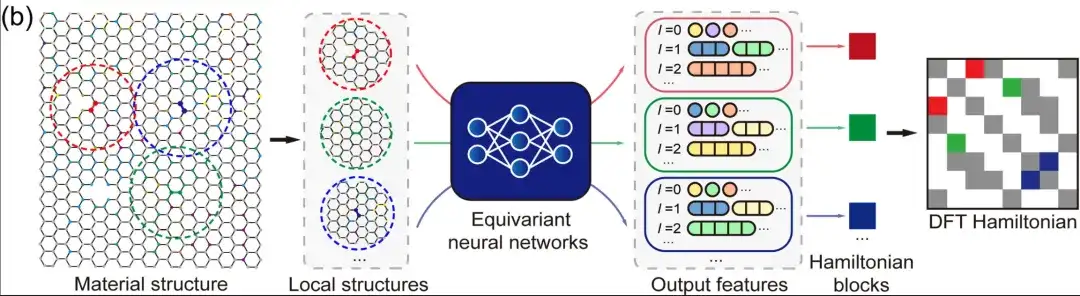
其次,在局域原子基组下,DFT 哈密顿量可以表示为稀疏矩阵,其矩阵元由局部化学环境决定。在等变神经网络 (Equivariant neural networks) 中,DeepH 利用不同角量子数 l 标记的输出特征来表示 DFT 哈密顿量,如上图所示。因此,人们可以根据临近结构信息来建模原子对之间的哈密顿量矩阵元,而不需要对整个材料结构的 DFT 哈密顿量矩阵进行建模。这不仅大大简化了深度学习任务,而且极大地增加了训练数据量。在推理方面,一旦深度学习网络学习到足够多的训练数据,经过训练的模型就可以很好地推广到更多未被见过的新材料结构。
DeepH 的关键思想是利用神经网络来表示 HDFT 。通过改变输入的物质结构,首先创建的是由 DFT 代码生成的 HDFT 训练数据,然后将这些数据用于训练神经网络。这些经过训练的网络模型随后再被用来对新的物质结构进行推理。
在这个过程中,存在两个非常重要的先验知识——其一是局部性原则,该研究在局域原子样本中表示 DFT 哈密顿量,并将哈密顿量分解为描述原子间耦合或原子内耦合的块。因此,单个训练材料结构可能对应于大量数据的哈密顿量块。此外,每个哈密顿量块可以根据局部结构的信息而不是整个结构来确定。这种简化确保了 DeepH 模型的高精度和可转移性。
其二是对称性原理,当从不同的坐标系观察时,物理定律保持不变。因此,相应的物理量和方程在坐标变换下表现出等价性。保持等价性不仅提高了数据效率,而且增强了泛化能力,这可以显著提高 DeepH 的性能。第一代 DeepH 架构通过局部坐标系简化了等价问题,并通过局部坐标的变换恢复了等价特征。第二代 DeepH 架构基于等价神经网络,名为 DeepH-E3 。在此框架中,所有输入、隐藏和输出层的特征向量都是等价矢量。最近,这项工作的作者之一提出了深度学习的新一代架构 DeepH-2 。在效率和准确性方面,DeepH-2 表现最优。
综上,该研究的深度学习模型 DeepH 使用 DeepH-2 方法训练,共包含 1,728 万个参数,基于 3 个等价变换块组成了可用于消息传递的神经网络,每个节点和边缘携带 80 个等价特征。材料结构的嵌入包含原子序数和原子间距离,采用高斯平滑策略,基函数的中心范围从 0.0 一直到 9.0Å。神经网络的输出特征则通过线性层传递,然后通过 Wigner-Eckart 层构建 DFT 哈密顿量。
该研究在 NVIDIA A100 GPU 上进行训练,共进行了 343 个时期,耗时 207 小时。在整个训练过程中,batch 大小固定为 1,这意味着每个 batch 包含一个材料结构。最后,初始学习率为 4×10-4,衰减速率为 0.5,衰减耐心为 20,最小选择的学习率为 1×10-5,并在学习率达到此值时停止训练。
DeepH 推理性能优异,可提供准确的能带结构预测
在训练、验证和测试集上,模型预测的密度泛函理论哈密顿量矩阵元的平均绝对误差 (MAE) 分别达到 1.45 、 2.35 和 2.20 meV,这表明该模型具有对未曾见过的结构进行推理的能力。
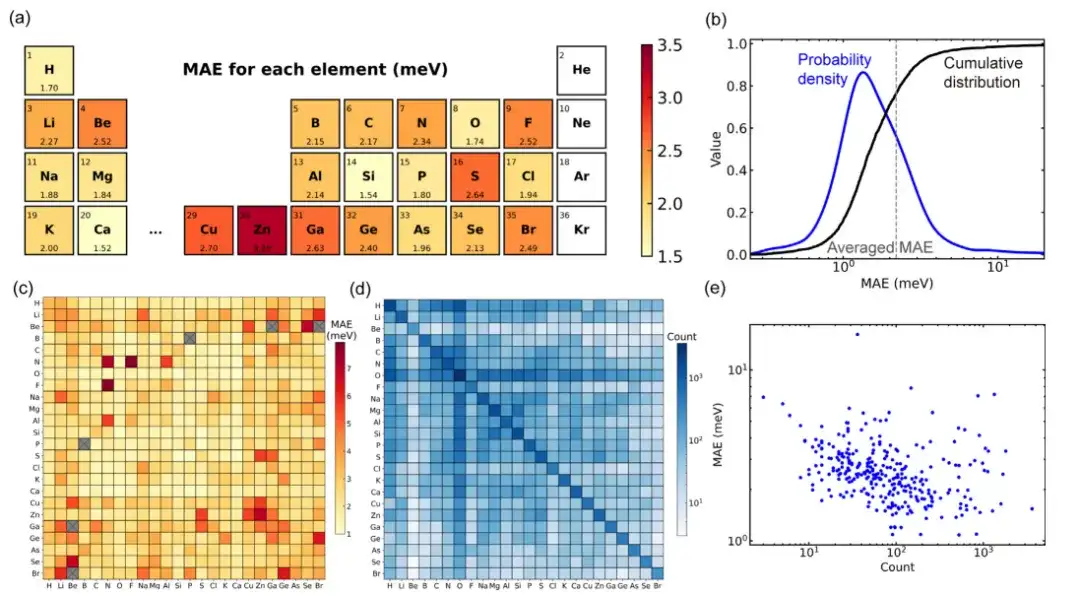
在利用 104 种固体材料的大型材料数据库对 Deep-2 方法训练的通用材料模型进行性能评估时,在数据集的所有结构中,大约 80% 的材料结构具有小于平均值 (2.2 meV) 的平均绝对误差。只有 34 个结构(约占测试集的 1.4%)的平均绝对误差超过 10meV,说明该模型对主流结构有良好预测精度。
通过进一步分析数据集,模型在材料结构上的性能偏差可能是由于数据集分布偏差造成的。研究发现,数据集中包含的元素对的训练结构越多,相应的平均绝对误差就越小。这一现象可能表明深度学习通用材料模型存在「缩放法则」,即更大的训练数据集或许会提高模型性能。
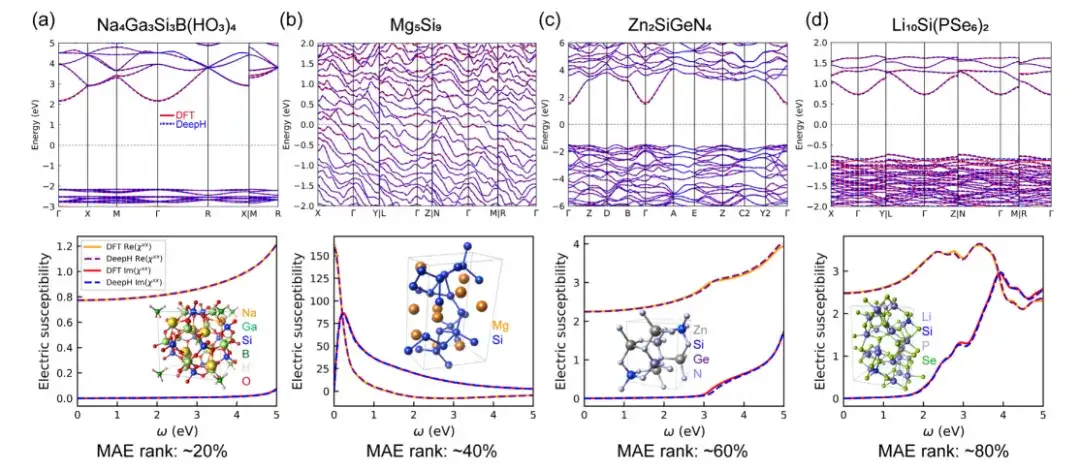
为了评估 DeepH 通用材料模型预测材料性质的准确性,该研究在计算示例时,分别使用了基于密度泛函理论 (DFT) 计算和 DeepH 预测的 DFT 哈密顿量,然后将这两种方法得到的计算结果进行了比较。结果表明,DeepH 预测的结果与 DFT 计算的结果非常接近,证明了 DeepH 在计算材料性质方面的出色预测精度。
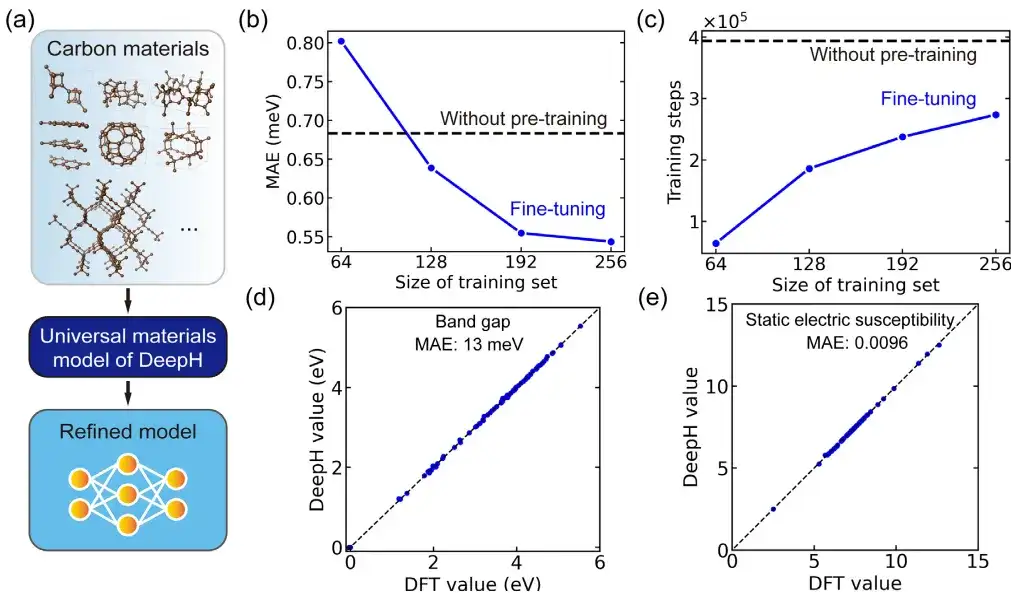
在具体的应用中,该研究用微调通用材料模型对碳同素异形体进行了研究。其中,碳材料数据集来源于萨马拉碳同素异形体数据库 (SACADA),共包含 427 种具有不同原子结构的碳同素异形体。
研究人员基于此对通用材料模型进行微调,创建了一个专门针对碳材料的改进型 DeepH 模型。与无预训练模型相比,微调可以将预测 DFT 哈密顿量的平均绝对误差显著降低至 0.54 meV,还可以在少于 50% 的训练结构中实现可比较的预测精度。
此外,微调还显著改善了训练收敛,并减少了训练时间。可以说,微调有助于提高预测准确性并增强训练效率。更重要的是,微调后的 DeepH 模型在预测材料性质方面表现出了显著的优势,经过微调的模型几乎可以为所有测试结构提供准确的能带结构预测。
材料大模型风起云涌, AI4S 任重道远
以 ChatGPT 为时间起点,AI 正式进入了一个全新的「大模型时代」。这个时代的特点是利用庞大的数据集和先进的算法,训练出能够处理复杂任务的深度学习模型。在材料科学领域,这些大模型正与研究者们的智慧相结合,开启了一个前所未有的研究新纪元。这些大模型不仅能够处理和分析海量的科学数据,还能够预测材料的性质和行为,从而加速新材料的发现和开发,推动着这一领域向更高效、更精准的方向发展。
在过去的一段时间,AI for Science 正在与材料科学不断碰撞出新的火花。
立足国内,北京凝聚态物理国家研究中心 SF10 组、中科院物理研究所、中科院计算机网络信息中心共同合作,将数万个化学合成路径数据投喂给大模型 LLAMA2-7b,从而获得了 MatChat 模型,可用来预测无机材料的合成路径;电子科技大学联合复旦大学、中国科学院宁波材料技术与工程研究所,成功开发出「耐疲劳铁电材料」,在全球范围内率先攻克困扰领域内 70 多年的铁电材料疲劳问题;上海交通大学 AIMS-Lab 实验室开发出了新一代材料智能设计模型 Alpha Mat.……研究成果频出,材料创新和发现进入新时代。
放眼全球,Google 旗下的 DeepMind 开发了用于材料科学的人工智能强化学习模型 GNoME,寻找到了 38 万余个热力学稳定的晶体材料,相当于「为人类增加了 800 年的智力积累」,极大加快了发现新材料的研究速度;微软发布的材料科学领域人工智能生成模型 MatterGen,可根据所需要的材料性质按需预测新材料结构;Meta AI 与美国高校合作,开发了行业顶级的催化材料数据集 Open Catalyst Project,以及有机金属框架吸附数据集 OpenDAC……科技巨头凭借自家的技术,将材料科学领域搅动的风起云涌。
虽然与传统材料研发方式相比,人工智能为探索更广泛的材料可能性打开了大门,显著减少了与材料发现相关的时间与费用。但是,AI for Science 在材料领域还面临着可信度和有效实施的挑战,确保数据质量、识别和减轻用于训练 AI 系统的数据潜在偏差等一系列问题有待解决。这或许也意味着,要想让人工智能在材料科学领域发挥更大的作用,仍然前路漫漫。