Command Palette
Search for a command to run...
韩国版 AlphaFold?深度学习模型 AlphaPPIMd:用于蛋白质-蛋白质复合物构象集合探索

在生命的舞台上,蛋白质扮演着不可或缺的角色。它们是生物体中最为活跃的分子,参与细胞的构建、修复、能量转换、信号传递以及无数关键的生物学功能。同时,蛋白质的结构与其功能密切相关,而它们的功能又通过与蛋白质、多肽、核苷酸以及各种小分子的复杂相互作用来实现。这种蛋白质-蛋白质相互作用 (PPI) 是细胞内许多生物过程的核心,从细胞信号传导到免疫反应,再到细胞周期的调控,无不涉及 PPI 。
然而,人们目前对蛋白质三维结构及其相互作用特性的了解仍不够全面。传统的实验技术,如 X 射线晶体学和冷冻电镜,虽然能够提供高分辨率的蛋白质结构信息,但耗时且成本高昂,且在解析动态过程和低丰度蛋白质时面临挑战。这极大限制了人们对蛋白质功能和相互作用机制的深入理解,进而影响了药物设计和蛋白质工程的发展。
针对于此,延世大学王建民博士及其合作者通过将深度学习与生成式 AI 相结合,利用基于 Transformer 的生成神经网络学习探索蛋白质-蛋白质复合物的构象集合,从多个分子动力学 (MD) 轨迹中学习了影响蛋白质-蛋白质复合物构象和动力学机制的关键残基,并为蛋白质-蛋白质结合提供了机理性见解。

论文地址:
https://doi.org/10.1101/2024.02.24.581708
AlphaPPIMd 模型:基于分子动力学模拟,以自注意力机制为核心
研究团队 barnase-barstar 复合物轨迹集作为数据集。首先从蛋白质数据库 (Protein Data Bank, PDB) 中下载了 barnase-barstar 复合物的晶体结构,通过去除配体和结晶水,提取 A 链和 D 链作为初始复合物结构。然后,研究人员通过 AmberTools 中的 tleap 模块添加缺失的氢原子,并通过加入 Na+ 和 Cl- 离子进行中和,在 12Å 的 TIP3P 水分子周期边界盒中进行溶剂化。最后,利用 AmberTools 中的 tleap 模块和 AMBER ff14SB 力场编制了系统的拓扑和坐标文件。
随后,研究团队使用分子动力学模拟系统,通过 Langevin 积分器进行了 500 步典型的 NVT 模拟,使得能量最小化。然后,在 300K 下通过 10,000 步 NPT 模拟来进一步达到平衡状态,并使用粒子网络 Ewald 算法,计算远程静电的相互作用,将直接空间相互作用的截断值设为 1nm,仿真时间步长设置为 2fs,同时还设置了 SHAKE 算法来约束所有涉及氢原子的键的长度,随即进行了 6 次独立的 100ns 分子动力学模拟。所有模拟均采用 OpenMM 7.7 进行。
在完成分子动力学模拟后,研究团队基于 Transformer 构建了 AlphaPPIMd 模型,利用深度生成模型来捕捉传统分子动力学难以分析的蛋白质构象状态。 AlphaPPImd 框架的核心是自注意力机制,可以从 MD 轨迹中捕获影响蛋白-蛋白复合物构象的关键氨基酸残基对。
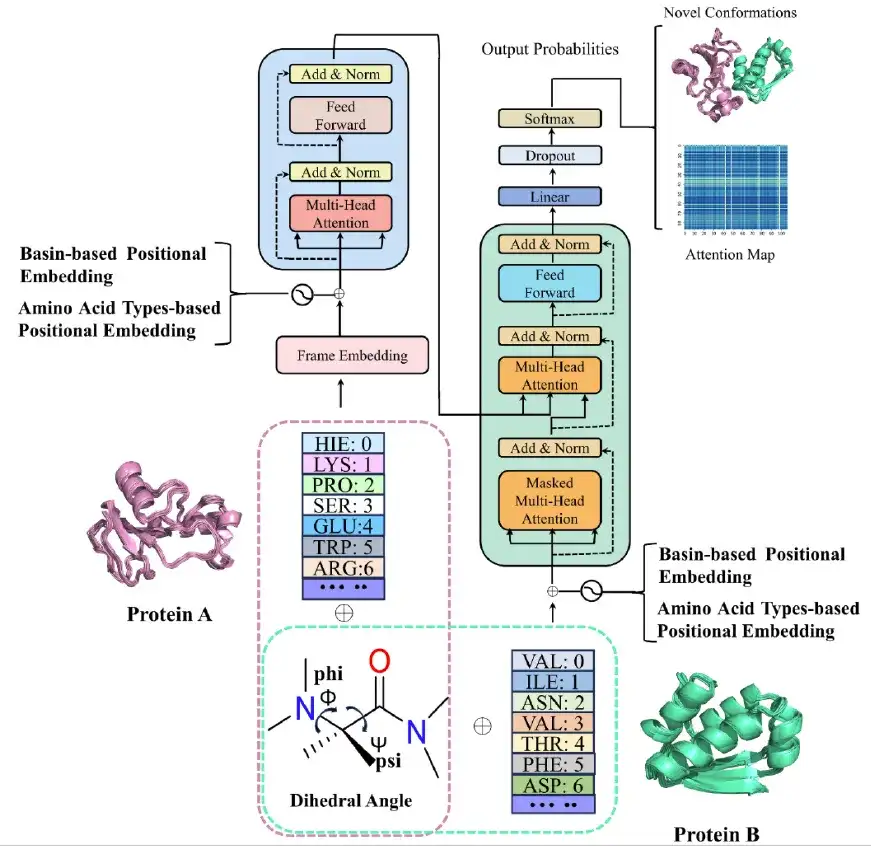
首先,AlphaPPImd 框架会对蛋白-蛋白复合物的 MD 轨迹进行预处理,得到两条链的序列长度、序列组成和氨基酸残基类型,并通过计算轨迹中选定残基的 Φ,Ψ 角度,以表示不同的构象状态。(如上图中粉色与绿色虚线框内所示)
其次,研究人员通过向量模块 (embedding module) 将蛋白-蛋白复合物 MD 轨迹的每一帧输入 AlphaPPImd 的编码器模块,该模块包含多头自注意力机制 (multi-head self-attention mechanism) 、注意力分数 (attention score) 和特征优化模块 ( feature optimization module) 。 AlphaPPImd 的解码器用于学习和捕获蛋白质复合物不同类型和位置的残基对构象的贡献。
最后,预测模块迭代生成下一帧的基态,Modeller 可根据扩展的基态编码轨迹重构蛋白质-蛋白质复合物的构象模型。
AlphaPPImd 解码器模块中的多头自注意层学习了特定残基对之间的相互作用,可以将注意力函数视为查询 (Q) 与键值对 (K-V) 输出之间的映射。 AlphaPPImd 采用蛋白质复合物残基嵌入作为 Q,将全局蛋白质复合物特征用作 K 和 V,并通过使用 Q 和 K 计算注意力权重。其计算公式如下:
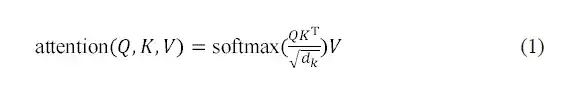
与此同时,该研究将 barnase-barstar 复合物的 6 个独立的 100ns MD 轨迹分为 300 个基元,每个基元由 1,000 帧组成。 MD 轨迹经过预处理,仅保留蛋白质原子。每个 MD 运行都提供了一组有限的蛋白质-蛋白质复合物物理快照。轨迹中的每一帧都表示为 Φ,Ψ 编码的基态。因此,蛋白质-蛋白质复合物的扭转状态被降维为一种文本表示形式,保留了动力学的主要次要特征。
研究结论:平均训练精度高达 0.995,可推广向更多蛋白质复合物
barnase-barstar 复合物由两条不同的链组成,共有 197 个残基组成 (barnase 链:108 个残基,barstar 链:89 个残基) 。该研究通过 KMeans 算法将点位划分为 4 个聚类,标记为 0(下图中的紫色)、 1(下图中的深蓝色)、 2(下图中的绿色)、 3(下图中的黄色),然后记录并存储每个簇的质心,以便从基态编码的扭转状态重建 barnase-barstar 复合物的全原子模型。
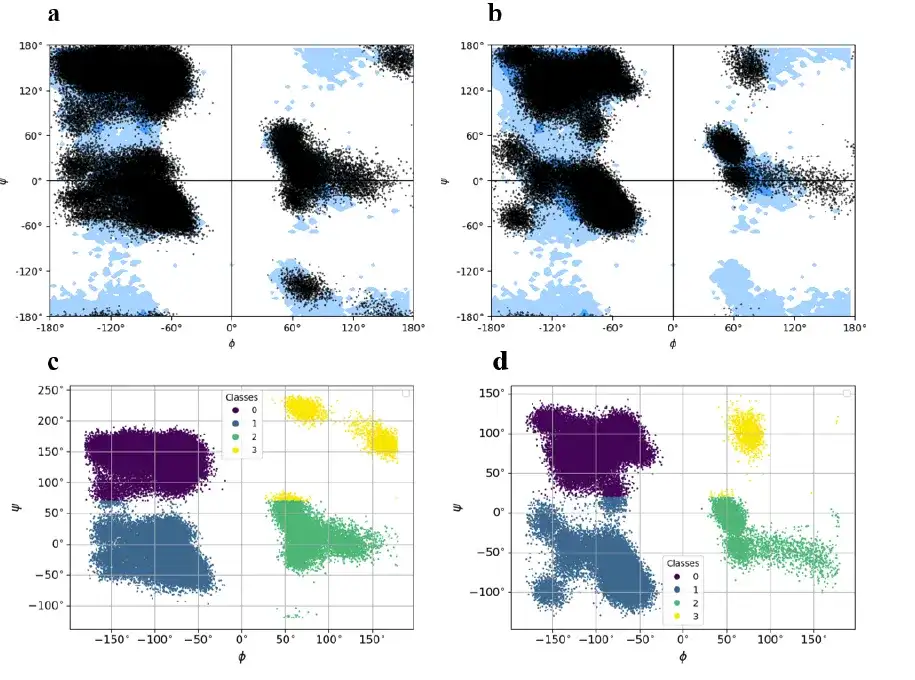
该研究将每一帧的轨迹转换成一个字符向量,每个字符向量由对应于 4 个簇的 4 个符号组成。最后,该研究对 barnase-barstar 复合体的 MD 轨迹数据集中的所有 300 个基元,执行了类似的表示过程。
综上所述,barnase-barstar 复合物是一种异二聚体,两条链中编码的残基基态差异明显。这意味着,barnase-barstar 复合物在生成新的基态编码框架,以及重建单个蛋白质的构象模型方面存在显著差异。
研究表明,AlphaPPImd 模型的平均训练精度为 0.995,平均验证精度为 0.999 。虽然 AlphaPPImd 很快就获得了稳定的性能,但为了进一步完善 Transformer 模型并丰富模型学习到的 MD 构象分布,该研究使用了多个 MD 轨迹作为数据集。例如,该研究通过从测试集的轨迹中随机选取一帧作为输入,并利用训练后的 AlphaPPImd 框架生成 100 个基态编码帧。
结果表明,该模型能够成功地对构象进行采样和展开,并且能够正确执行 Φ、Ψ 的二面体约束。
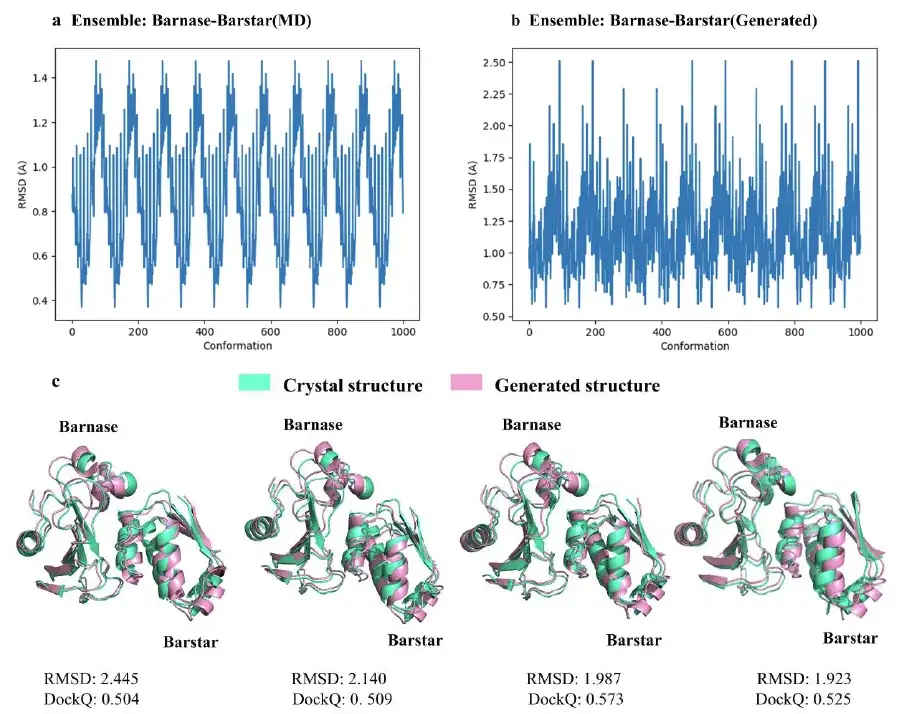
该研究还从 AlphaPPImd 模型生成的 1,000 个 barnase-barstar 复合物构象中,选择了 4 个 RMSD 接近 2Å 的代表性构象。研究结果表明,AlphaPPImd 生成的蛋白质复合物构象模型与参考晶体结构更接近,准确度更高(均方根偏差 < 2Å)且可接受性更高(DockQ ≥ 0.23)。
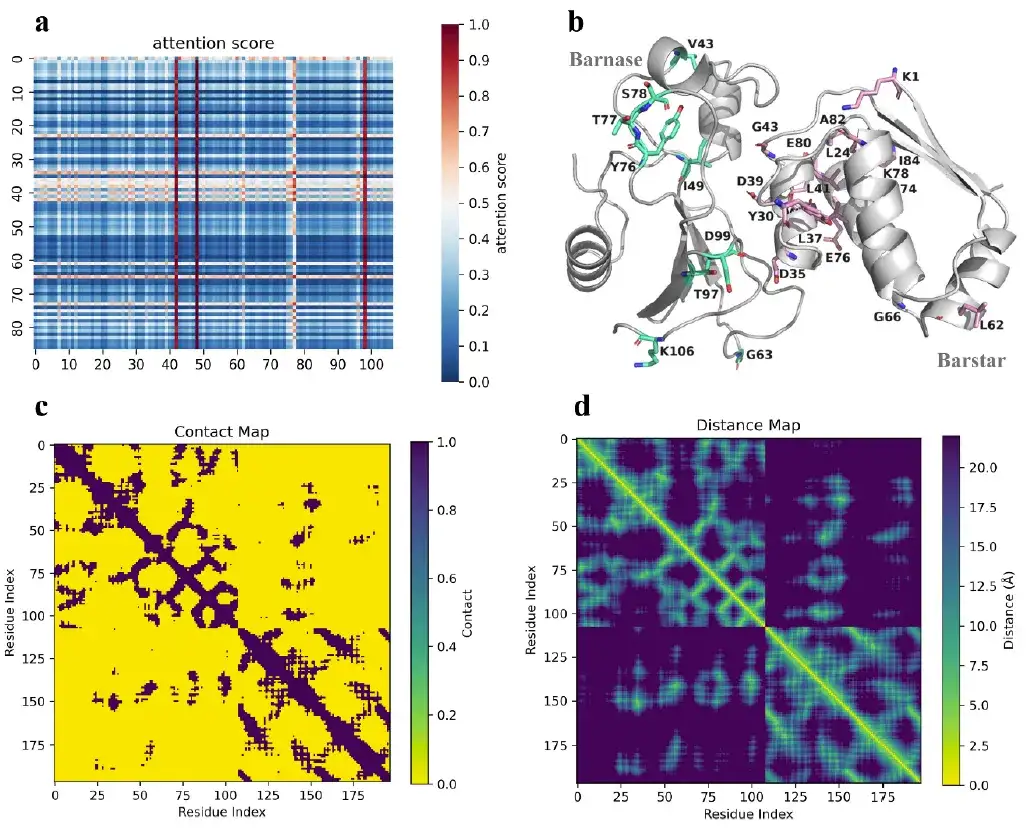
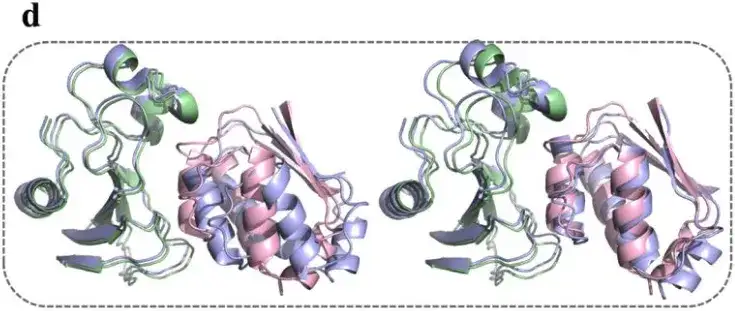
此外,AlphaPPImd 的注意力机制捕获了关键残基之间的注意力权重,并提供了关于蛋白质-蛋白质结合的机制见解。
研究表明,AlphaPPImd 模型捕获的关键残基主要位于蛋白质相互作用、环和螺旋的接口处,这意味着,深度生成模型从 barnase-barstar 复合物的 MD 轨迹中捕获了影响其动力学和构象的关键残基,可用于补充 MD 结果。同时,AlphaPPImd 模型捕获的关键残基主要位于 Mdm2-p53 相互作用界面,这也证明该模型可以推广到其他蛋白质-蛋白质复合物。
AI 蛋白质预测:从 AlphaFold 到百家争鸣
早在 2016 年,AlphaGo 名声大噪之后,DeepMind 团队就开始了针对蛋白质折叠问题的研究。
在 2018 年底的第 13 届 CASP (Critical Assessment of protein Structure Prediction) 中,AlphaFold 在 98 名参赛者中名列榜首,准确地从 43 种蛋白质中预测出了 25 种蛋白质的结构。 2020 年,AlphaFold 2 面世,实现了蛋白质单体结构的高准确度预测。 2021 年 10 月,DeepMind 发布了一个名为 AlphaFold-Multimer 的更新,其基于 AlphaFold 2 进行了拓展,可以对多种蛋白质的复合物进行建模。 2024 年 5 月 8 日,AlphaFold 3 再次惊艳世人,将预测范围从蛋白质带到广泛的生物分子。
早在 AlphaFold 2 推出之际,中科院院士施一公就曾对媒体表示:「依我之见,这是人工智能对科学领域最大的一次贡献,也是人类在 21 世纪取得的最重要的科学突破之一,是人类在认识自然界的科学探索征程中一个非常了不起的历史性成就。」
有了 AlphaFold 的珠玉在前,AI 在蛋白质设计领域引发的产业革命悄然而至。
2023 年,全球首个 AI 蛋白质生成大模型 NewOrigin(中文名「达尔文」)在世界制造业大会上正式亮相。据介绍,NewOrigin 大模型基于条件生成机制,联合使用 AI 、分子动力学、量子计算、湿实验等多维反馈机制,可高精度生成蛋白质序列、蛋白质功能、蛋白质知识表示等多种模态蛋白质内容,完成亲和力、稳定性、活性、表达量等多维度任务,满足真实的产业应用所需。
2022 年,华盛顿大学医学院的生物学家在 Science 上连发两篇论文,介绍了他们的重大发现。研究者表示,使用机器学习可以在几秒钟内创建出蛋白质分子。而在以前,这个时间会长达几个月。创造出自然界中没有的蛋白质,有助于疫苗研发、加快治疗癌症的研究、碳捕获工具研发、可持续生物材料研发等。
毫无疑问,AI 蛋白结构预测能够让我们更好地认识和理解蛋白质,进而理解生命。但是,仅仅只是认识和理解是远远不够的,科学家们未来需要通过 AI 预测蛋白质来解决医疗领域的实际问题,例如按需求改造蛋白质、甚至从头设计自然界不存在的蛋白质。道阻且长,期待 AI 能够在生命科学探索中带来更多惊喜。