Command Palette
Search for a command to run...
LAMMPS: 단결정 알루미늄을 예로 들어 재료의 단축 인장 시뮬레이션
LAMMPS(Large-scale Atomic/Molecular Massively Parallel Simulator)는 재료 모델링에 중점을 둔 고전적인 분자 동역학 시뮬레이션 코드입니다. 병렬 컴퓨터에서 효율적으로 실행되고 쉽게 확장 및 수정할 수 있도록 설계되었습니다. LAMMPS는 원래 미국 에너지부 산하 기관인 샌디아 국립 연구소에서 개발되었으며, 현재는 여러 연구 그룹과 여러 기관의 전문가들의 기여를 받고 있습니다. LAMMPS 개발 자금의 대부분은 미국 에너지부(DOE)에서 지원받습니다. LAMMPS는 GNU Public License 버전 2(GPLv2)에 따라 배포되는 오픈 소스 소프트웨어입니다.
1. 튜토리얼 콘텐츠
이 튜토리얼에서는 재료의 격자 상수를 변경하여 재료에 단축 변형률을 가하는 상황을 시뮬레이션하고, 재료의 변형률-응력 곡선을 계산하여 그립니다. 이 튜토리얼을 통해 다음 내용을 배우게 됩니다.
- npt를 사용하여 시스템 구조를 완화하세요
- 격자 상수를 변경하려면 변수 명령과 함께 fix를 사용하세요.
- 시스템 스트레스를 계산하려면 compute 명령을 사용하세요.

2. 입력 파일
- Al99.eam.합금 재료의 EAM 잠재력
- in.txt 입력 파일
- pl.py 그림 이미지 스크립트
3. 입력 파일 파싱
시스템 초기화 및 기본 설정
units metal # 采用金属单位制(长度:Å,时间:ps,质量:g/mol,能量:eV 等)
dimension 3 # 三维空间模拟
boundary p p p # 三个方向均为周期性边界条件(模拟无限大晶体)
atom_style atomic # 原子类型为「基本原子」,仅包含位置和类型信息
variable latparam equal 4.05 # 定义铝的晶格常数为 4.05Å(FCC 铝的典型值)
원자 구조 구축
lattice fcc ${latparam} orient x 1 0 0 orient y 0 1 0 orient z 0 0 1 # 定义 FCC 晶格 # 晶格常数为 ${latparam},晶向沿 x[100] 、 y[010] 、 z[001](标准笛卡尔坐标系)region whole block 0 10 0 10 0 10 # 定义模拟区域:一个立方体,在 x 、 y 、 z 方向各包含 10 个晶格常数长度
create_box 1 whole # 创建容纳原子的「盒子」,仅包含 1 种原子类型,边界为上述 region 定义的范围
create_atoms 1 region whole # 在盒子内填充类型为 1 的原子,按 FCC 晶格排列
원자간 상호작용 전위 설정
pair_style eam/alloy # 采用嵌入原子法(EAM)合金势描述原子间相互作用(适合金属体系)
pair_coeff * * Al99.eam.alloy Al # 配置势函数参数:
# - 第一个*:所有原子类型(此处仅 1 种)
# - 第二个*:所有原子类型的相互作用
# - Al99.eam.alloy:势函数文件(预训练的铝势)
# - Al:指定原子类型 1 对应元素为铝
계산 볼륨 정의(후속 분석을 위해)
compute csym all centro/atom fcc # 计算每个原子的中心对称参数(用于识别 FCC 结构完整性,缺陷处会偏离)
compute peratom all pe/atom # 计算每个原子的势能(用于分析能量分布)
평형 시뮬레이션 단계(초기 응력 제거 및 안정 상태 도달)
reset_timestep 0 # 重置时间步计数器为 0(平衡阶段作为新起点) timestep 0.001 # 时间步长为 0.001ps(即 1fs,金属模拟常用精度) #初始化原子速度:温度 300K(室温),随机种子 12345,消除整体动量(mom yes)和旋转(rot no)
velocity all create 300 12345 mom yes rot no #施加 NPT 系综(恒温、恒压、恒粒子数)进行平衡:
#- temp 300 300 1:温度维持 300K,热浴阻尼系数 1(单位:ps)
#- iso 0 0 1:各向同性压力控制,目标压力 0bar,压力阻尼系数 1(单位:ps)
#- drag 1:原子运动阻尼系数(用于稳定系综)
fix 1 all npt temp 300 300 1 iso 0 0 1 drag 1 #热力学输出设置:
thermo 1000 #每 1000 步输出一次热力学信息
thermo_style custom step lx ly lz press pxx pyy pzz pe temp #输出内容:
#步数、盒子尺寸(x/y/z)、总压力、压力张量分量、势能、温度 run 20000 #运行 20000 步平衡模拟(总时间 20000×0.001ps=20ps)
unfix 1 #平衡结束,移除 NPT 约束#记录平衡后的 x 方向盒子长度(用于后续计算应变)
variable tmp equal "lx" #临时变量存储当前 x 方向长度
variable L0 equal ${tmp} #定义 L0 为初始长度(拉伸前的平衡长度)
print "Initial Length, L0: ${L0}" #输出初始长度,便于验证
단축 인장 변형 단계
reset_timestep 0 #重置时间步计数器为 0(拉伸阶段作为新起点) #重新施加 NPT 系综,但仅控制温度和横向压力:
#- temp 300 300 1:维持温度 300K
#- y 0 0 1 z 0 0 1:y 和 z 方向压力保持 0bar(横向自由,模拟单轴拉伸)
fix 1 all npt temp 300 300 1 y 0 0 1 z 0 0 1 drag 1 #定义拉伸速率:
variable srate equal 1.0e10 #应变速率为 1e10 /s(分子动力学常用高应变速率,远高于实验)
variable srate1 equal "v_srate / 1.0e12" #转换为 LAMMPS 单位(应变速率单位:ps⁻¹,1e10/s = 1e-2 ps⁻¹)#施加 x 方向拉伸变形:
#- deform 1 x:沿 x 方向变形,变形组为 1(所有原子)
#- erate ${srate1}:按上述应变速率拉伸
#- units box:变形基于盒子尺寸
#- remap x:原子坐标随盒子拉伸同步更新(避免原子「跑出」盒子)
fix 2 all deform 1 x erate ${srate1} units box remap x
스트레칭 프로세스 데이터 출력
#定义应变和应力变量: variable strain equal "(lx - v_L0)/v_L0" #工程应变 =(当前 x 长度 - 初始长度)/ 初始长度 variable p1 equal "v_strain" #p1 对应工程应变(无量纲) variable p2 equal "-pxx/10000" #x 方向应力(GPa):pxx 为 LAMMPS 内置压力张量(单位 bar),负号转为拉应力,除以 10000 转换为 GPa variable p3 equal "-pyy/10000" #y 方向应力(GPa) variable p4 equal "-pzz/10000" #z 方向应力(GPa) #输出应变-应力数据到文件:
#- 每 100 步输出一次 p1(应变)、 p2(x 应力)、 p3(y 应力)、 p4(z 应力)
#- 保存到 Al_tens_100.def1.txt,不在屏幕显示
fix def1 all print 100 "${p1} ${p2} ${p3} ${p4}" file Al_tens_100.def1.txt screen no #输出原子轨迹文件(用于可视化):
#- 每 100 步保存一次,文件名为 md.lammpstrj
#- 包含原子 ID 、类型、 x/y/z 坐标
dump trace all custom 100 md.lammpstrj id type x y z #热力学信息输出设置:
thermo 1000 #每 1000 步输出一次
thermo_style custom step v_strain temp v_p2 v_p3 v_p4 ke pe press #输出步数、应变、温度、三方向应力、动能、势能、总压力run 30000 #拉伸阶段运行 30000 步(总时间 30ps,累积应变约 0.3)
시뮬레이션 종료
print "All done" # 输出完成信息,提示模拟结束
4. 시뮬레이션 시작하기
1. 克隆本容器后进入容器设置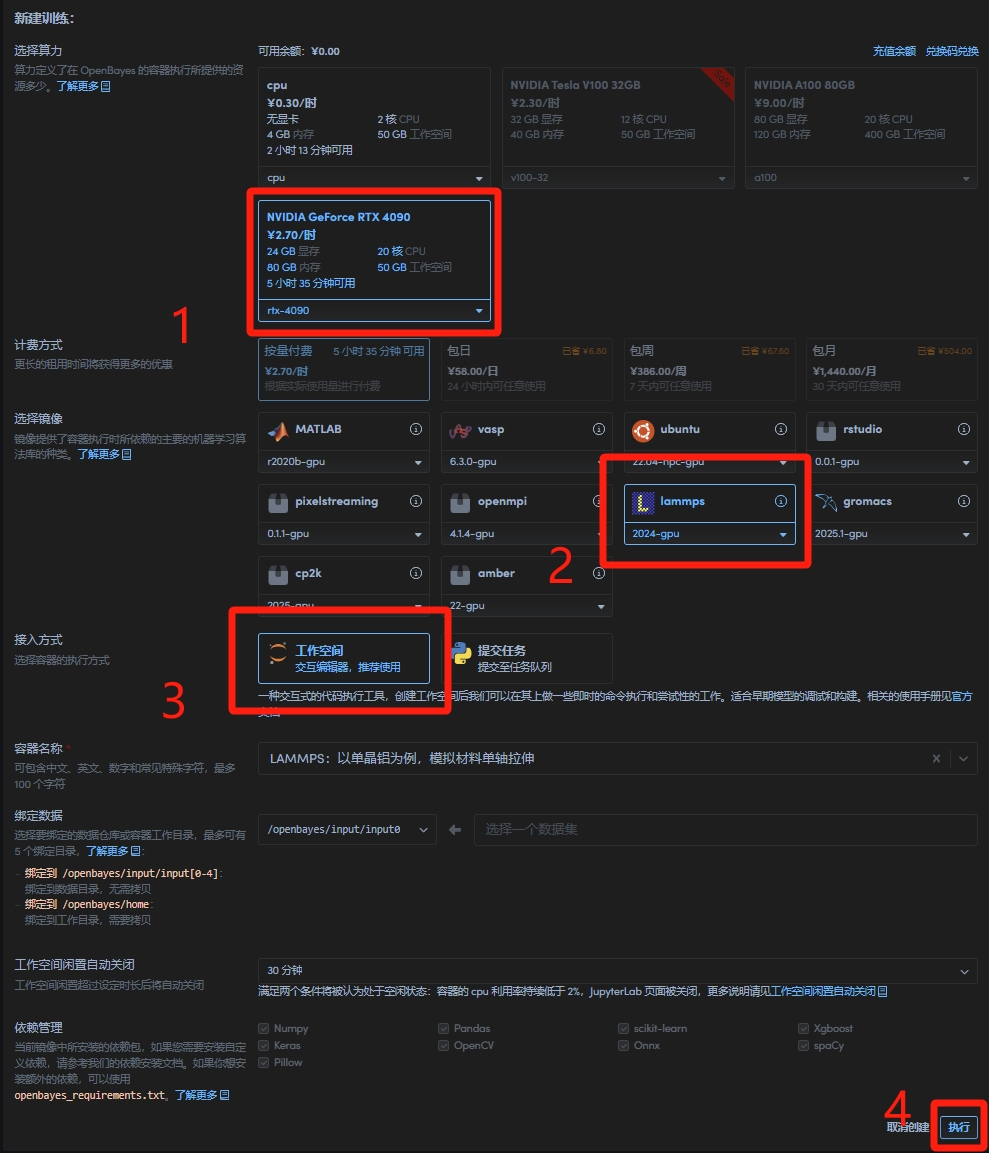
2. 待加载完毕后打开工作空间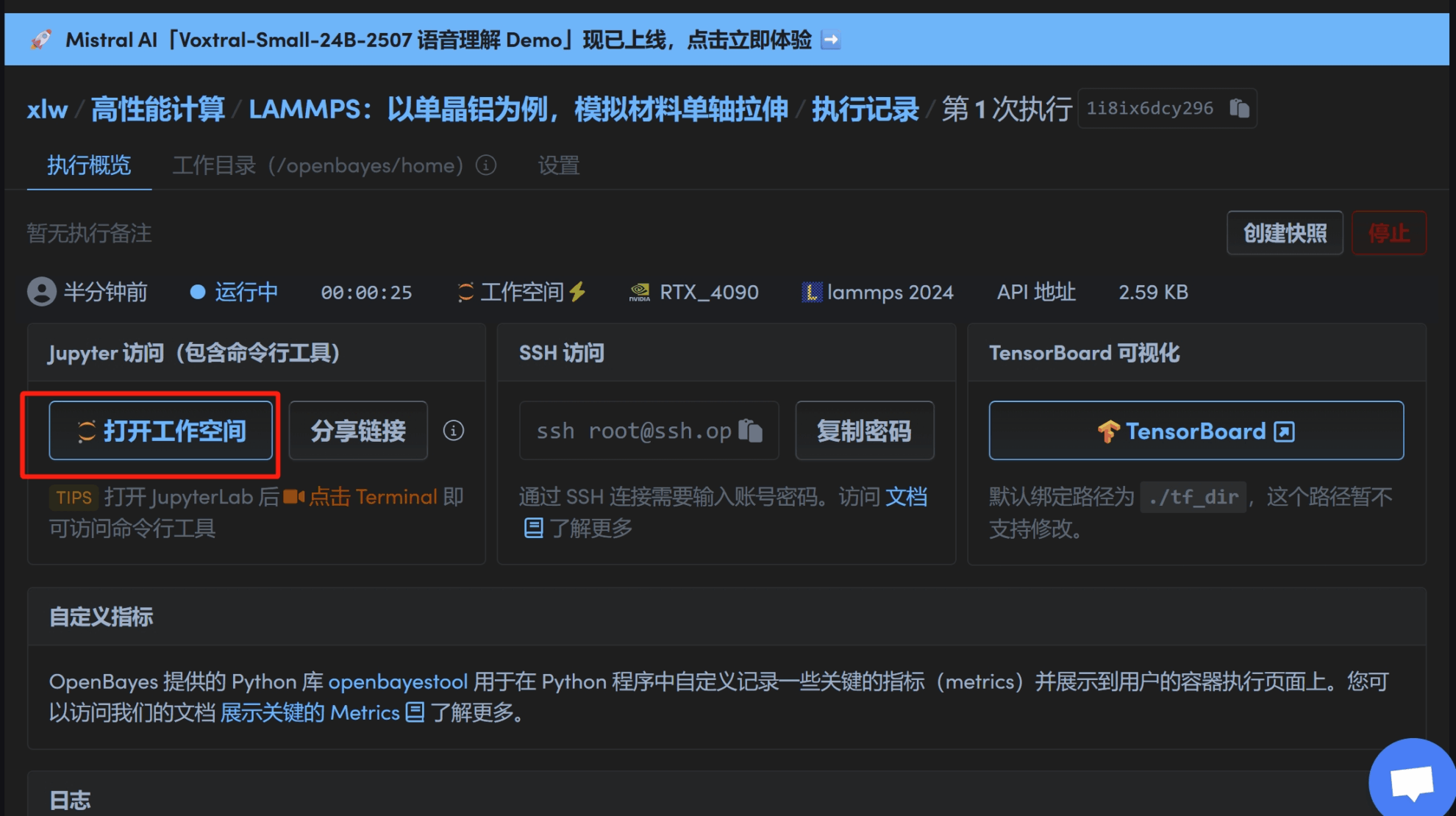
3. 随后打开终端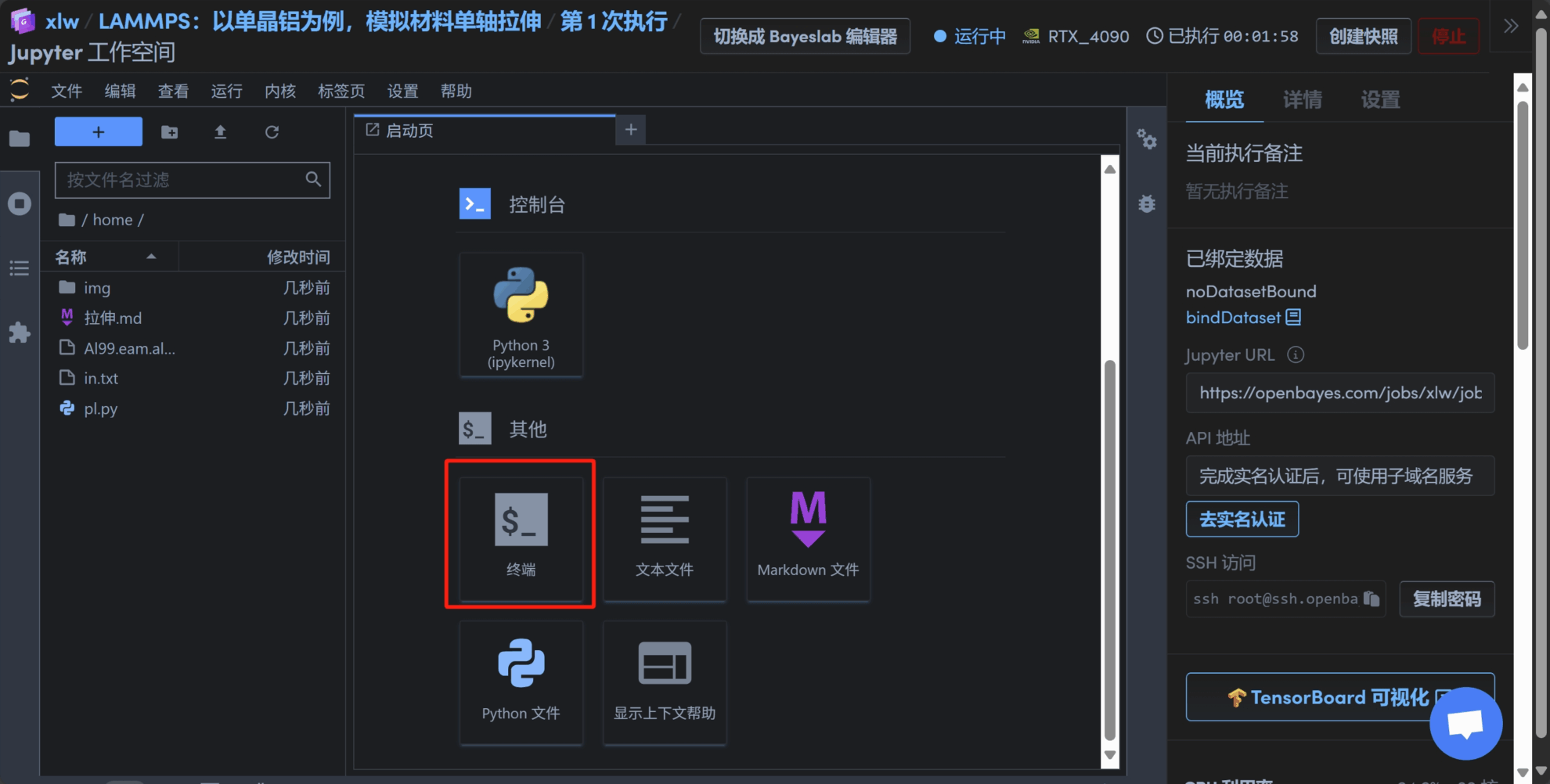
4. 输入以下命令运行 lammpsmpirun --allow-run-as-root -np 2 lmp -sf gpu -pk gpu 1 < in.txt 
5. 파일 출력
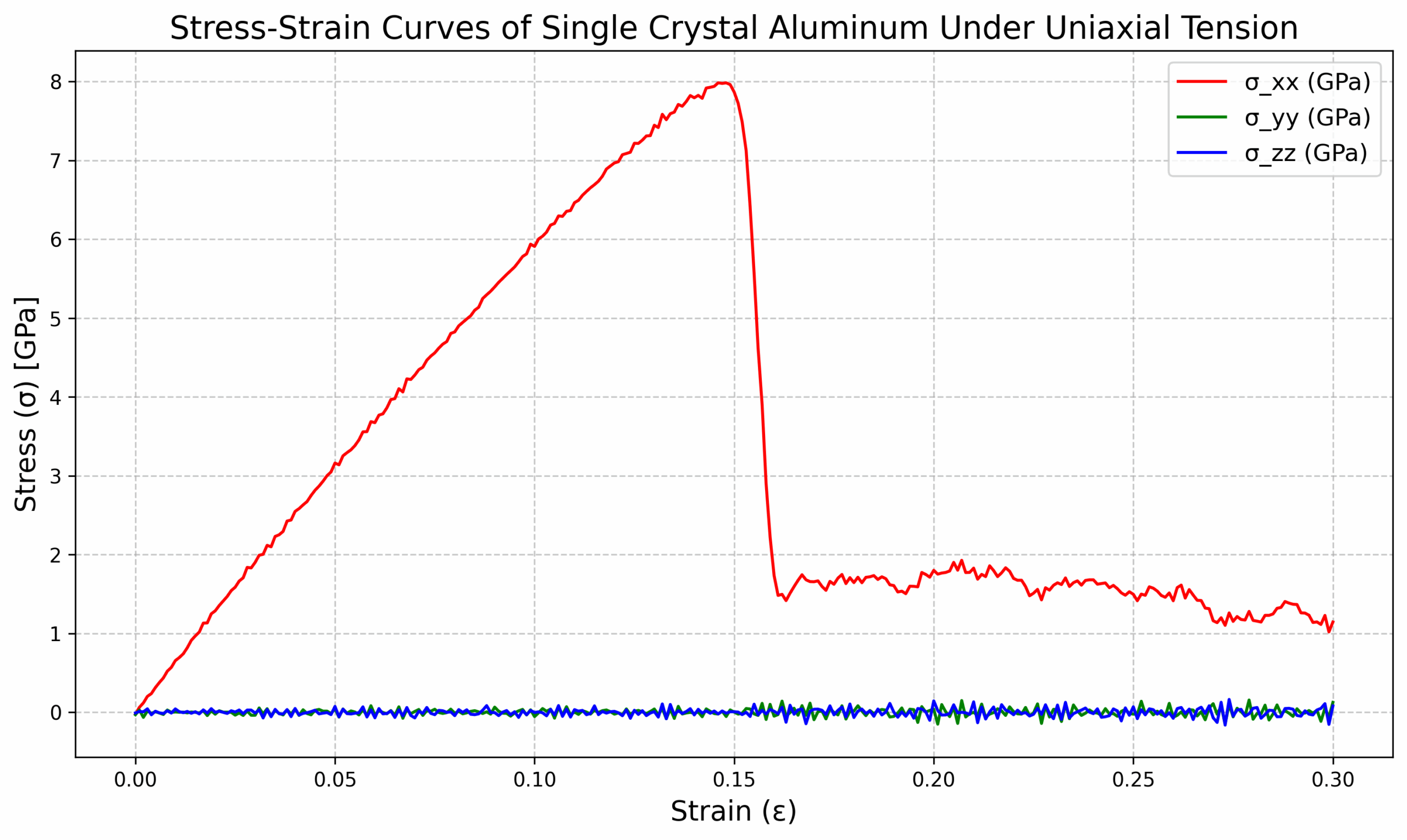
- Al_tens_100.def1.txt:변형률-응력 곡선 데이터는 탄성 계수, 항복 강도와 같은 기계적 특성을 분석하는 데 사용할 수 있습니다.
- md.lammpstrj: VMD 및 Ovito와 같은 소프트웨어를 통해 스트레칭하는 동안 결정 구조 진화를 시각화하는 데 사용할 수 있는 원자 궤적 파일
6. 변형률-응력 그래프 그리기
1. 在终端输入以下命令python pl.py2. 打开绘制的图片
3. 可以得到曲线