Command Palette
Search for a command to run...
단일 세포 전사체 시퀀싱 단일 샘플 튜토리얼: 품질 관리, 클러스터링, (차등) 유전자 표시
단일 세포 전사체 시퀀싱 분석 튜토리얼
이 샘플 데이터는 "진행성 간세포암에 대한 개인화된 신항원 백신과 펨브롤리주맙: 1/2상 시험"환자 6의 인간 말초혈액 단핵세포의 단일 세포 시퀀싱 데이터(Yarchoan M et al. Nat Med. 2024;30:1044–1053.)
단일 세포 시퀀싱 소개
단일 세포 시퀀싱은 과학자들이 개별 세포 수준에서 유전자 발현을 심층적으로 분석할 수 있게 해주는 정교한 생명공학입니다. 이 과정은 일반적으로 조직 샘플을 먼저 개별 세포로 분리하여 단일 세포 현탁액을 형성하는 것으로 시작됩니다. 다음으로, 오일-물 기술을 사용하여 각 세포를 개별적으로 캡슐화하여 후속 분석 중에 독립적으로 유지되도록 했습니다.
단일 세포 시퀀싱에서는 각 세포의 mRNA 분자에 고유한 분자 식별자를 표시합니다. 이 과정을 UMI(Unique Molecular Identifier) 라벨링이라고 합니다. 동시에 각 셀에는 동일한 바코드 태그가 지정되는데, 이 바코드 태그는 데이터 분석 단계에서 각 셀의 출처를 추적하는 데 사용됩니다. 이런 방식으로 연구자들은 각 세포의 mRNA 정보를 정확하게 측정하여 각 세포의 유전자 발현 상태를 이해할 수 있습니다.
1. 바코드 기능 : 셀 정보 기록
2. 단일 세포 시퀀싱에 UMI를 적용: 각 mRNA 분자에 대한 고유 식별자를 제공함으로써 유전자 발현 측정의 정확도를 향상시킬 뿐만 아니라 PCR 증폭 중 발생할 수 있는 오류를 줄여 전체 시퀀싱 프로세스의 신뢰성과 효과성을 향상시킵니다.
기본 프로세스
단일 세포 RNA 시퀀싱(scRNA-seq) 프로세스는 주로 단일 세포 분리 및 포획, 세포 용해, 역전사(RNA를 cDNA로 변환), cDNA 증폭 및 라이브러리 준비를 포함합니다.
이미지 출처: Kulkarni, A., et al. (2019). “대량을 넘어서: 단일 세포 전사체 분석 방법 및 응용에 대한 검토” Curr Opin Biotechnol 58: 129-136
실행 단계
이제 튜토리얼의 실제적인 부분으로 들어가겠습니다.
컨테이너를 시작하고 데이터를 업로드합니다.
1. 컨테이너를 시작하세요
다음 그림과 같이 컨테이너를 생성하고 rstudio를 열고 package-installed-gpu 버전을 선택한 다음 리소스가 할당된 후 R studio에 들어가기 위해 API 주소를 직접 엽니다(이 단계에서는 실명 인증이 필요합니다).
- 참고: 소프트웨어에 접속한 후 계정과 비밀번호는 rstudio입니다.
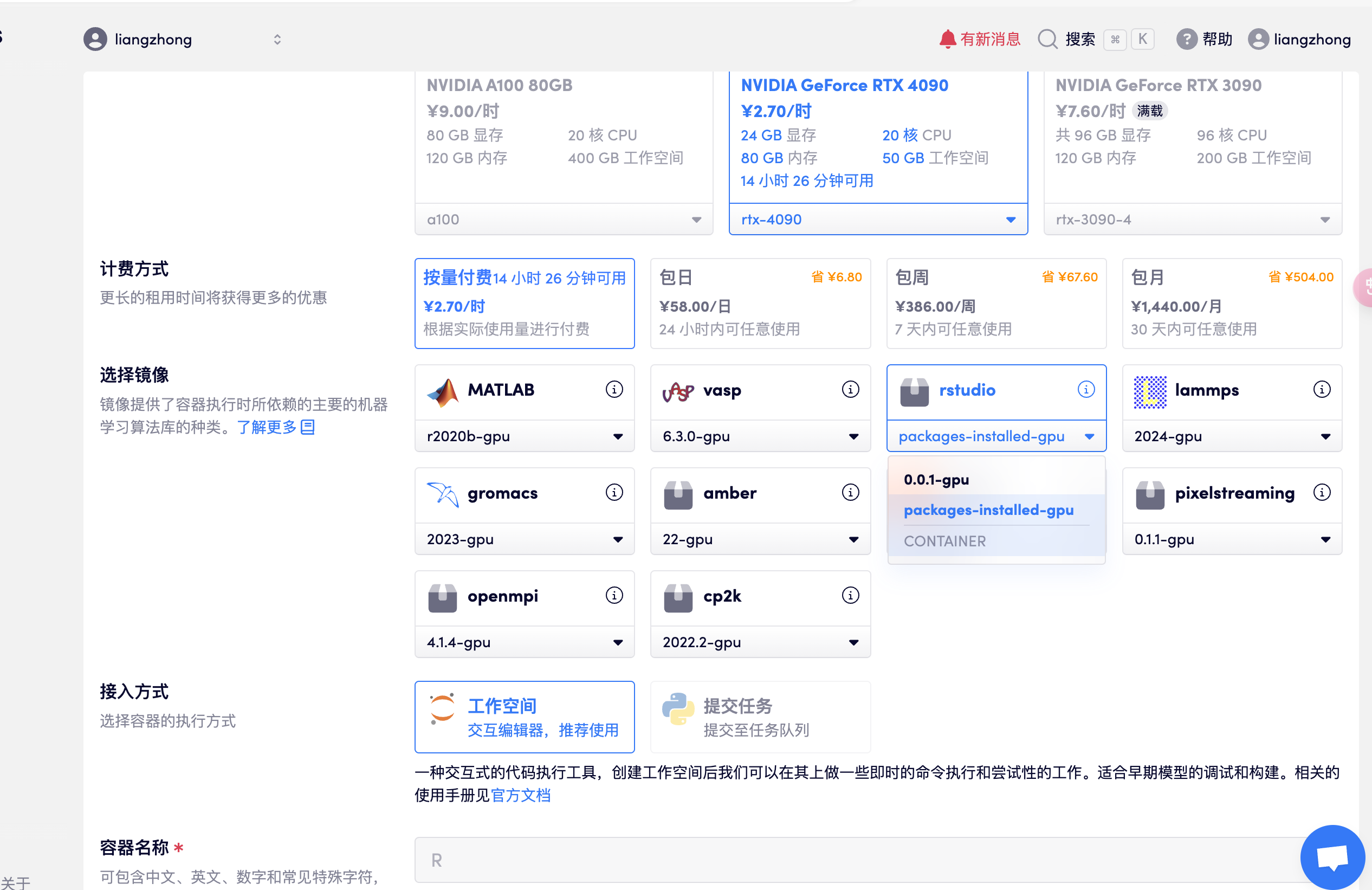
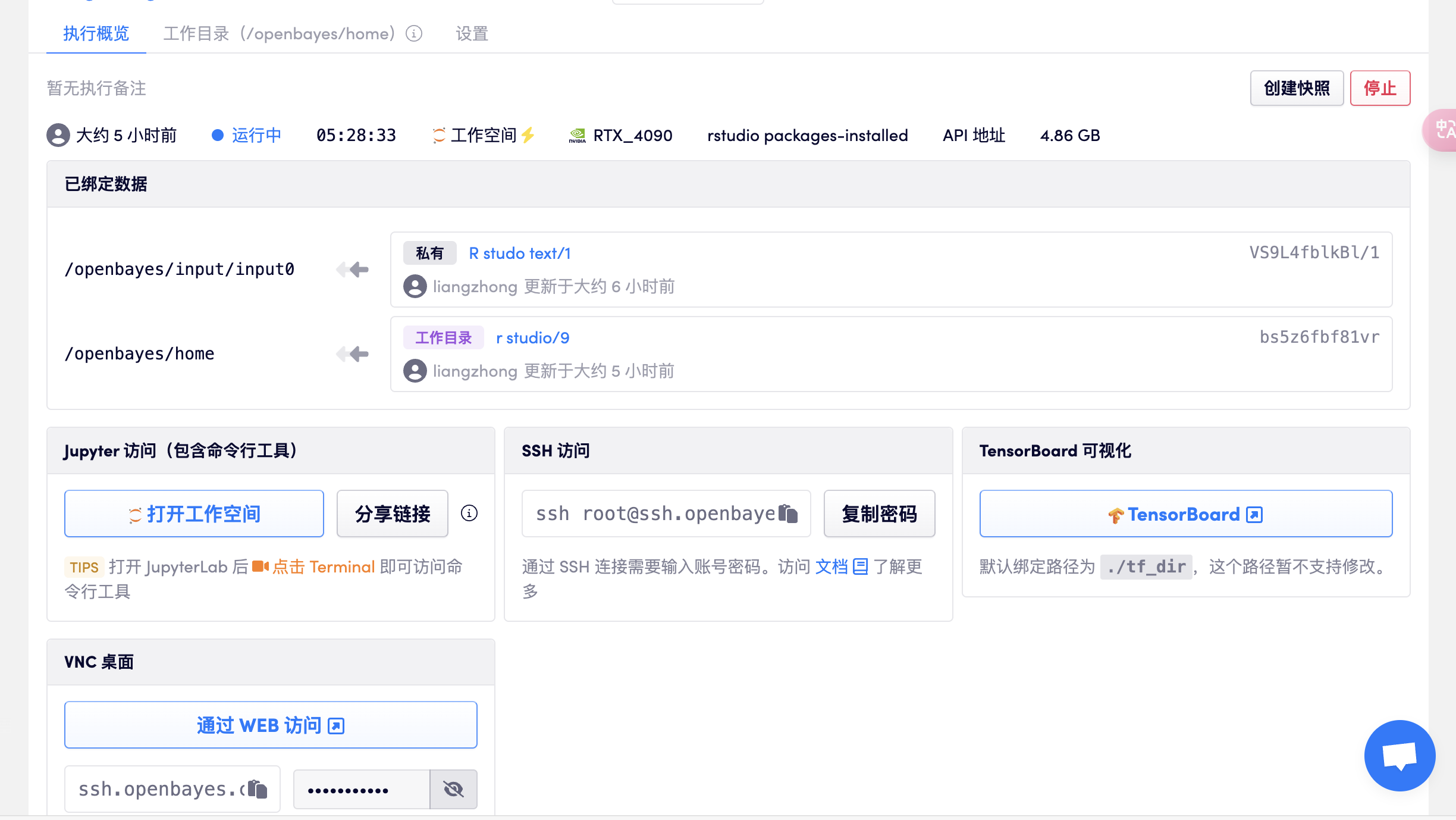
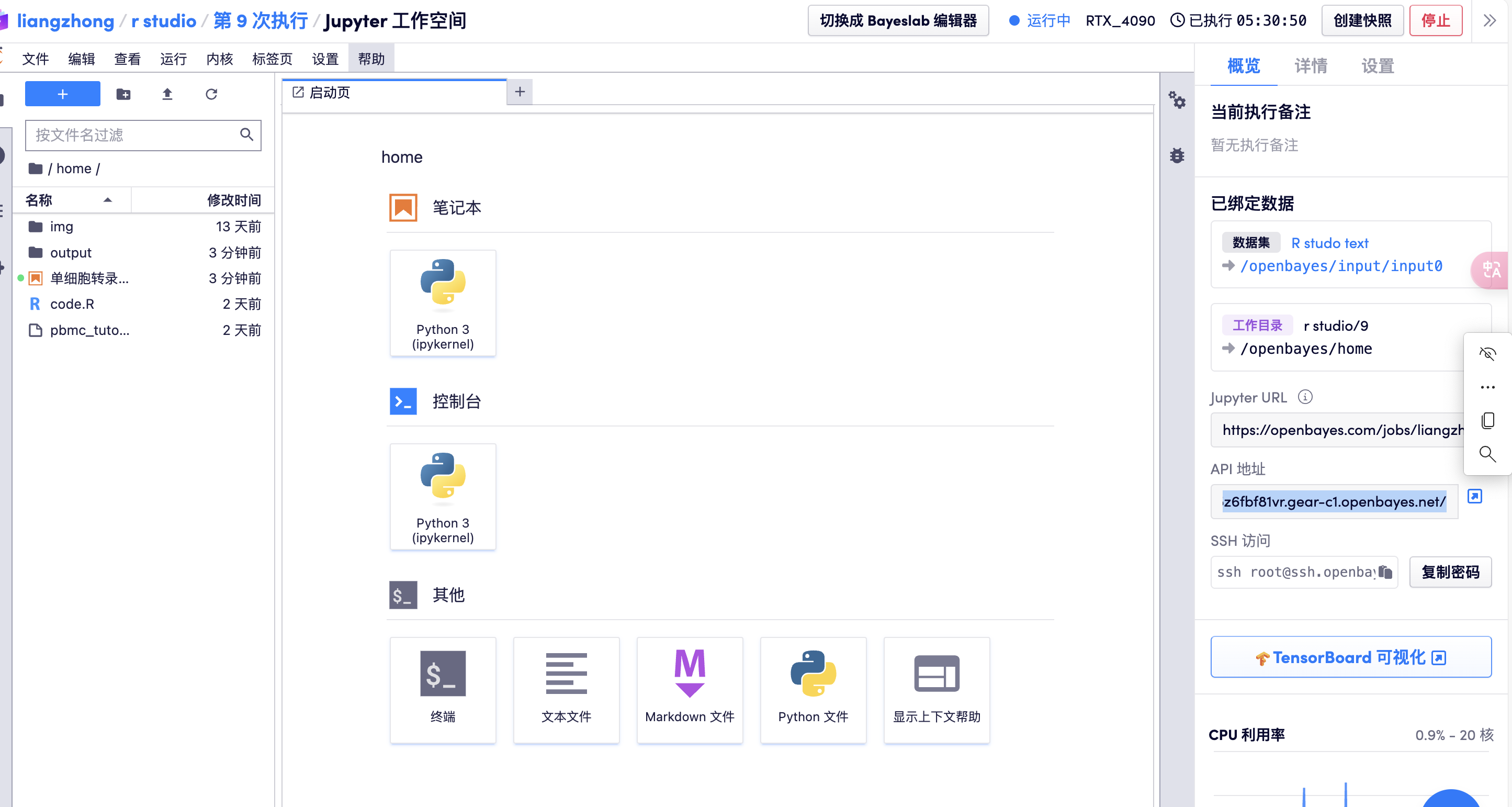
2. 데이터 업로드
미리 준비된 데이터를 업로드합니다(작업 디렉토리의 데이터 폴더에서 데이터를 다운로드하여 사용할 수 있습니다):
(1) Illumina NextSeq 500으로 시퀀싱한 단일 세포의 원시 데이터: barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz
(2) 셀 주석에 대한 참조 데이터: pbmc_multimodal.h5seurat, ref_Human_all.RData
#存放的目录如下
#此处可新建自定义文件夹,指定输入输出文件目录,本教程以 output 为例
rstudio@****(用户名)-bs5z6fbf81vrmain:~/home/output/data$ ls
barcodes.tsv.gz features.tsv.gz matrix.mtx.gz
rstudio@****(用户名)-bs5z6fbf81vr-main:~/home/output/reference_annotation$ ls
pbmc_multimodal.h5seurat ref_Human_all.RData다음으로, 튜토리얼 단계에 따라 단계별로 R 스튜디오에 코드를 입력합니다.
#报错改为英文
Sys.setenv(LANGUAGE = "en")
options(stringsAsFactors = FALSE)
#清空所有数据
rm(list=ls())
set.seed(100)#!!!设置随机数,保证后续实验的可重复性1. Seurat 객체 설정
# I. Setup -----------------------------------------------------------------------
# ## a. Load libraries ----------------------------------------------------------------------
```{r libraries}
library(Seurat)
library(SeuratObject)
library(sctransform)
library(patchwork)
library(Seurat)
library(SeuratData)
library(patchwork)
library(SeuratData)
library(ggplot2)
library(tidyverse)
library(glmGamPoi)
library(tools)
library(dplyr)
library(patchwork)2. 데이터 로딩
# II. Load data-----------------------------------------------------------------------
### a. Load data-----------------------------------------------------------------
getwd()#查看当前工作目录
setwd("~/home/output/results")
#修改工作目录,此处可根据自定义文件夹,指定输入输出文件目录,本教程以 output 为例
pbmc.data <- Read10X(data.dir ="~/home/output/data")#设置 10x 原始数据的工作目录,读取
seurat <- CreateSeuratObject(counts = pbmc.data, project = "pbmc", min.cells = 3, min.features = 200)#创建 seurat 对象
seurat
An object of class Seurat
16356 features across 10066 samples within 1 assay
Active assay: RNA (16356 features, 0 variable features)
1 layer present: counts
#该样本有 10066 个细胞 ,10066 个单细胞的 RNA 表达数据,测序了 16356 个基因
#counts: 通常表示原始的基因表达计数数据
colnames(seurat.final@meta.data)
[1] "orig.ident" "nCount_RNA" "nFeature_RNA" "percent.mt" "RNA_snn_res.0.1" "seurat_clusters"
[7] "celltype"
아래 그림은 pbmc.data 데이터를 보여줍니다. 읽기, 행 이름은 유전자, 열 이름은 세포 바코드입니다.
pbmc.data[c("CD3D", "CD8A", "PTPRC"), 1:30] "dgCMatrix" 클래스의 3 x 30 희소 행렬 [[ 30개 열 이름 'AAACCTGAGCGAAGGG-1', 'AAACCTGAGGCCCTTG-1', 'AAACCTGAGGGTTTCT-1' … 제외 ]]
CD3D 2 . 3 6 4 1 1 3 . 6 3 4 2 1 2 1 . 2 2 5 2 3 . 6. 4 3 CD8A. 6. . . 6 PTPRC 1 2 2 2 3 5 3 3 3 1 3 . 2 4 1 . 1. 2 9 . 3 4 . 1 2 . 2 1 4
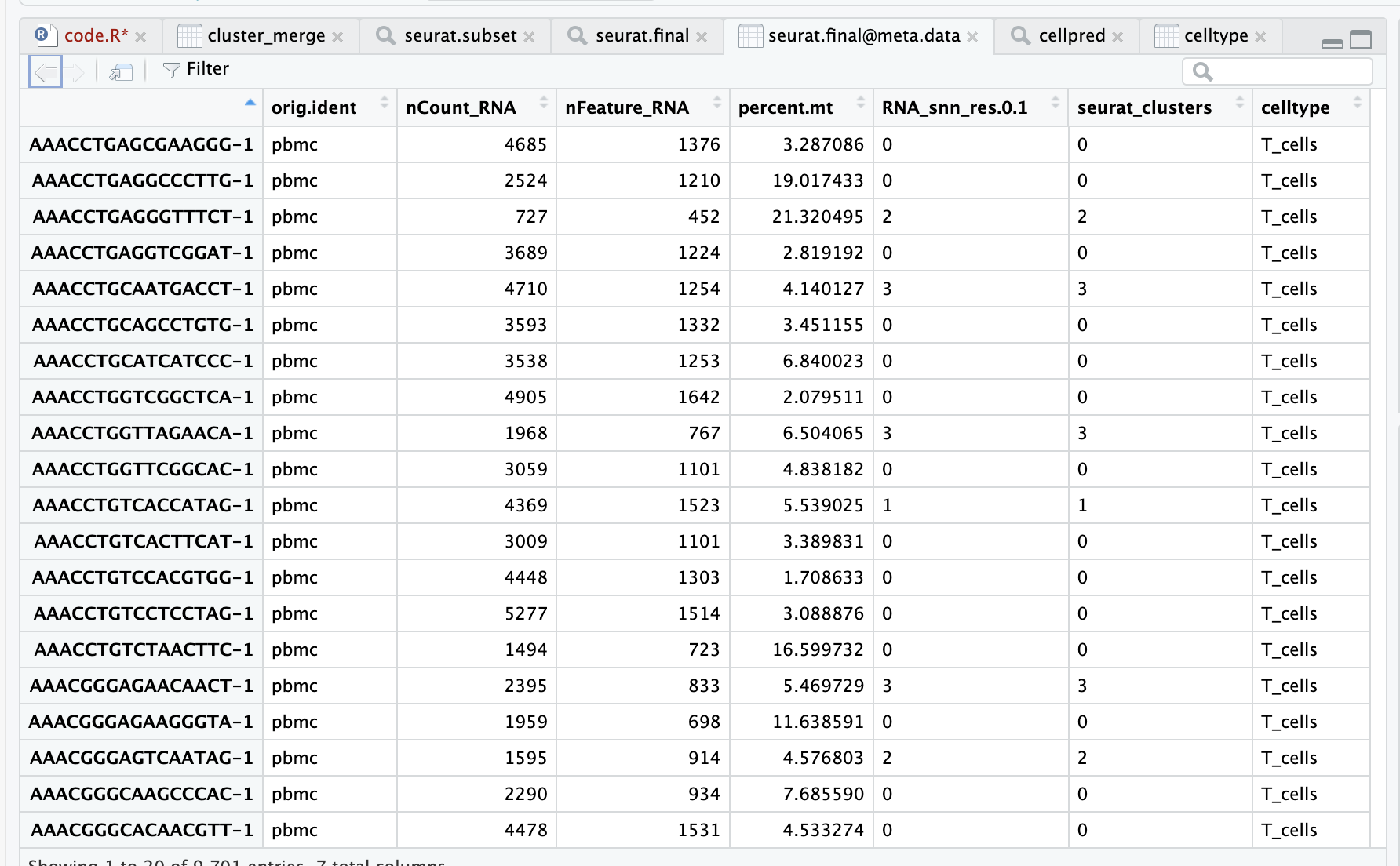
- 테이블 정보 설명:
- 행 이름은 바코드입니다
- orig.ident는 샘플 정보입니다.
- nCount_RNA는 총 유전자 발현량입니다.
- nFeature_RNA는 유전자의 수입니다.
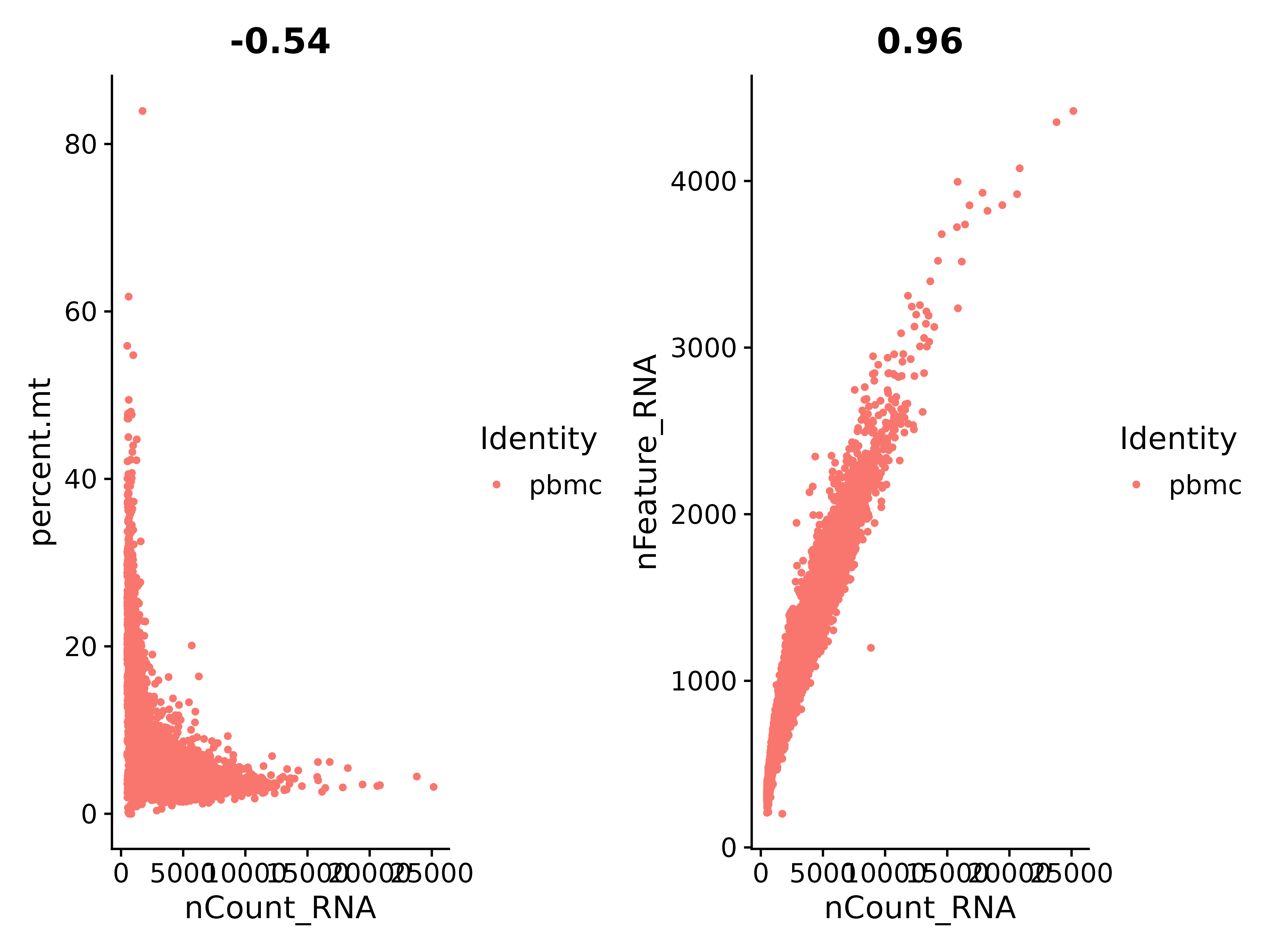
위 그림에 따라 nCount_RNA와 percent.mt(미토콘드리아 유전자)의 비율을 확인해 보겠습니다.
nCount_RNA와 nFeature_RNA는 0.96의 계수로 양의 비례관계를 가지고 있으며, 상관관계가 매우 좋은 것을 알 수 있습니다.
팁: MT-로 시작하는 모든 유전자는 미토콘드리아 유전자입니다.
3. 표준 전처리
다음 단계에서는 Seurat의 scRNA-seq 데이터에 대한 표준 전처리 워크플로를 다룹니다. 이러한 프로세스에는 QC 지표를 기반으로 셀을 선택하고 필터링하고, 데이터 정규화 및 크기 조정을 하고, 변동성이 큰 기능을 감지하는 작업이 포함됩니다.
Seurat는 사용자가 정의한 기준에 따라 QC 지표를 보고 셀을 필터링할 수 있습니다.
# III. Pre-processing----------------------------------------------------------------
### a. Plots for percent mito, nFeature---------------------------------------------
```{r preprocessing}
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
seurat <-PercentageFeatureSet(seurat, pattern = "^MT-", col.name = "percent.mt")
# Use FeatureScatter is typically used to visualize feature-feature relationships, but can be used
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt",)
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
ggsave("质控_1.png", plot1 + plot2, width = 8, height = 6, dpi=600)
#plot3 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.ribo")
#plot3
다음으로, QC 지표를 시각화하고 이를 사용하여 셀을 필터링합니다.
미토콘드리아 유전자의 비율을 살펴보고 바이올린 플롯을 사용하여 QC 지표를 시각화합니다.
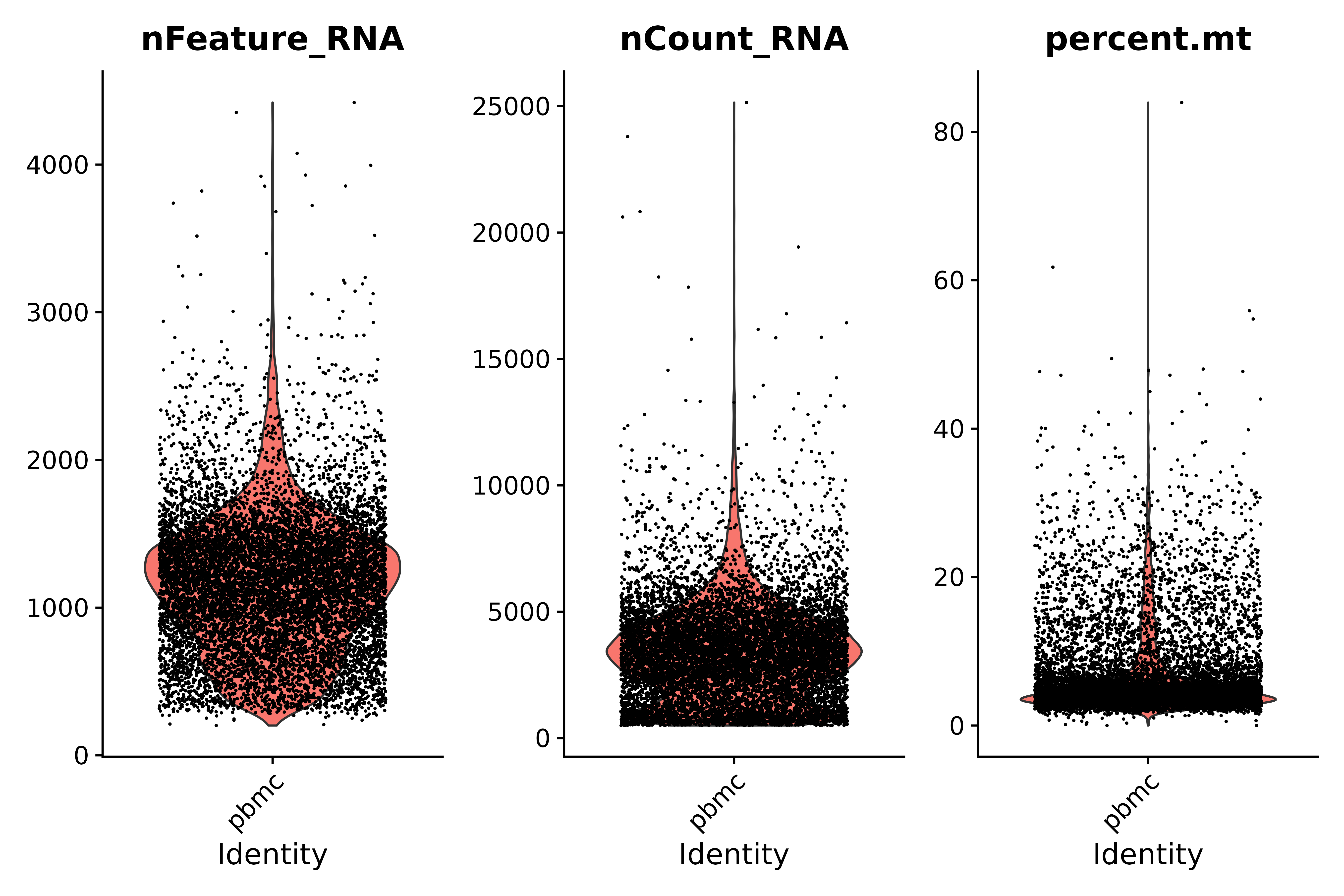
- 품질 관리 지침:
- 품질이 낮은 세포나 빈 물방울은 일반적으로 유전자가 거의 없습니다. nFeature_RNA > 200 & nCount_RNA > 500
- 그래프에 따르면 이배체 또는 다배체 세포는 비정상적으로 높은 유전자 수를 보일 수 있습니다. nFeature_RNA < 2500 & nCount_RNA > 500
- 품질이 낮거나 죽어가는 세포는 일반적으로 광범위한 미토콘드리아 과형성을 보이며, 백분율은 25 미만입니다.
# Visualize QC metrics as a violin plot
vln1 <- VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"#,"percent.ribo"
),ncol=3)
vln1
ggsave("质控_2.png", vln1, width = 9, height = 6, dpi=600)seurat.subset <- subset(seurat, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & nCount_RNA > 500 & nCount_RNA < 10000 &percent.mt < 25)
vln3 <- VlnPlot(seurat.subset, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
vln3
ggsave("质控_3.png", vln3, width = 9, height = 6, dpi=600)
pdf("qc_plots.pdf");plot1 + plot2;vln1;vln3;dev.off()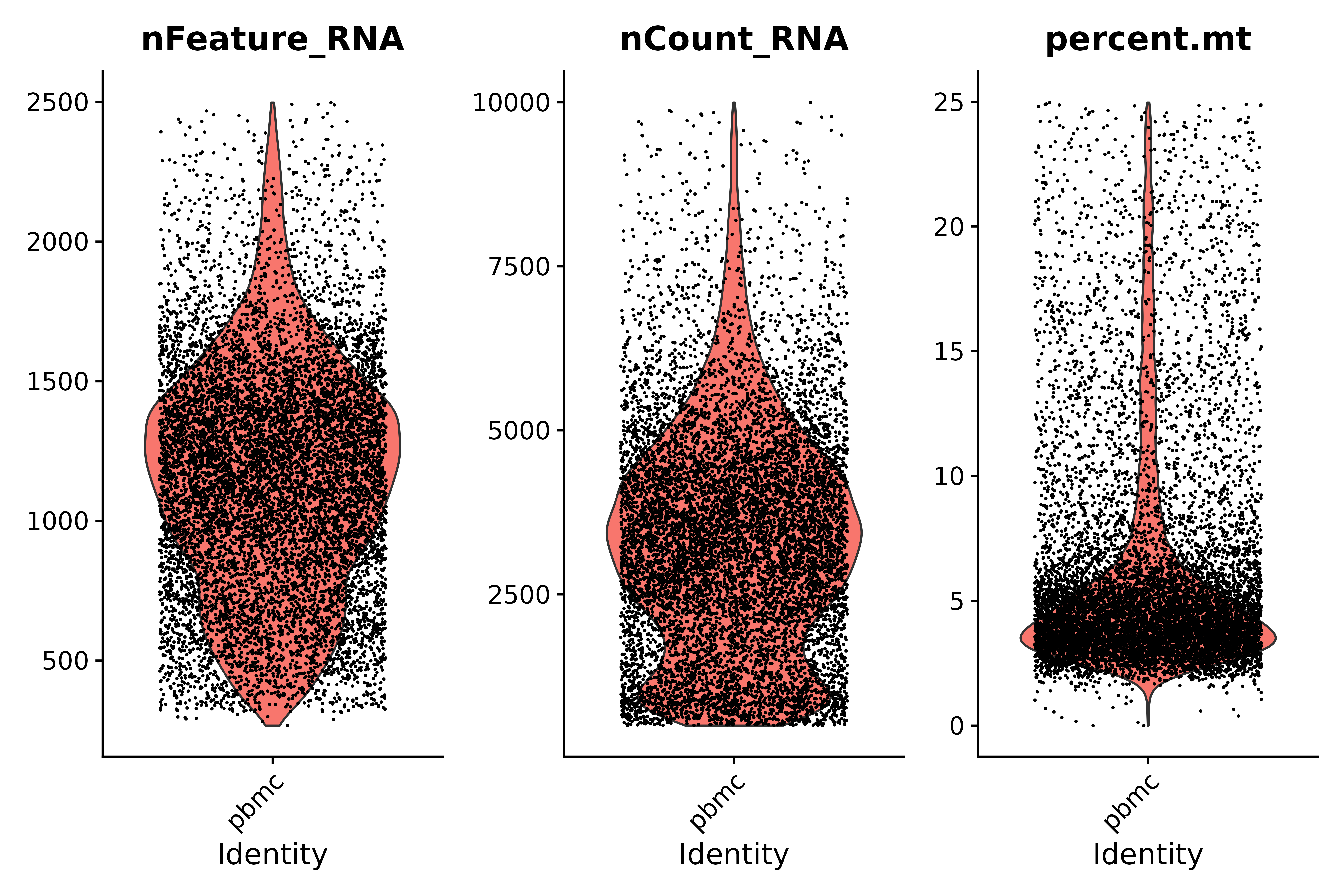
4. 데이터 정규화 및 변동성이 큰 기능 식별(기능 선택)
단일 세포 전사체 데이터 분석에서는 먼저 데이터 정규화를 수행하여 서로 다른 세포 간의 유전자 발현 수준을 효과적으로 비교할 수 있는지 확인합니다.
1. 데이터 정규화
- 데이터 정규화: 먼저 각 세포의 특정 유전자의 발현 수준(개수)을 세포의 총 발현 수준(총개수)으로 나눕니다. 이 단계는 세포 간의 시퀀싱 깊이 차이를 제거하여 서로 다른 세포의 유전자 발현 수준을 동일한 기준에서 비교할 수 있도록 하는 것입니다.
- 스케일링 조정: 정규화된 발현 값에 스케일링 계수(여기서는 10000)를 곱하여 후속 분석에 더 적합한 크기로 유전자 발현 값을 조정합니다.
- 대수 변환: 마지막으로 조정된 발현 값에 대해 대수 변환을 수행했는데, 이를 통해 분산을 더욱 안정화하고 발현량이 높은 유전자와 발현량이 낮은 유전자 간의 차이를 더 명확하게 보여 후속 분석 단계의 기초를 마련하는 데 도움이 되었습니다.
- 정규화: 위의 단계를 거쳐 유전자 발현 값을 10,000개 UMI 수준으로 정규화했습니다. 이는 세포 간 시퀀싱 깊이 차이의 영향을 제거하는 데 도움이 됩니다.
2. 다음으로, 돌연변이 특징을 검색합니다.
정규화와 표준화를 거친 후, 유전자 발현의 변동성을 분석하여 고가변 유전자를 식별했습니다. 이러한 유전자는 세포마다 발현 수준이 상당히 다르며, 일부 세포에서는 높게 발현되는 반면 다른 세포에서는 낮은 수준으로 발현될 수 있습니다.
# IV. Normalize Data----------------------------------------------------------------
seurat.subset <- NormalizeData(seurat.subset, normalization.method = "LogNormalize", scale.factor = 10000)
seurat.subset <- FindVariableFeatures(seurat.subset, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(seurat.subset), 10)
# plot variable features with and without labels
plot3 <- VariableFeaturePlot(seurat.subset)
plot4 <- LabelPoints(plot = plot3, points = top10, repel = TRUE)
ggsave("VariableFeaturePlot_4.png", plot3 + plot4, width = 12, height = 6, dpi=600)
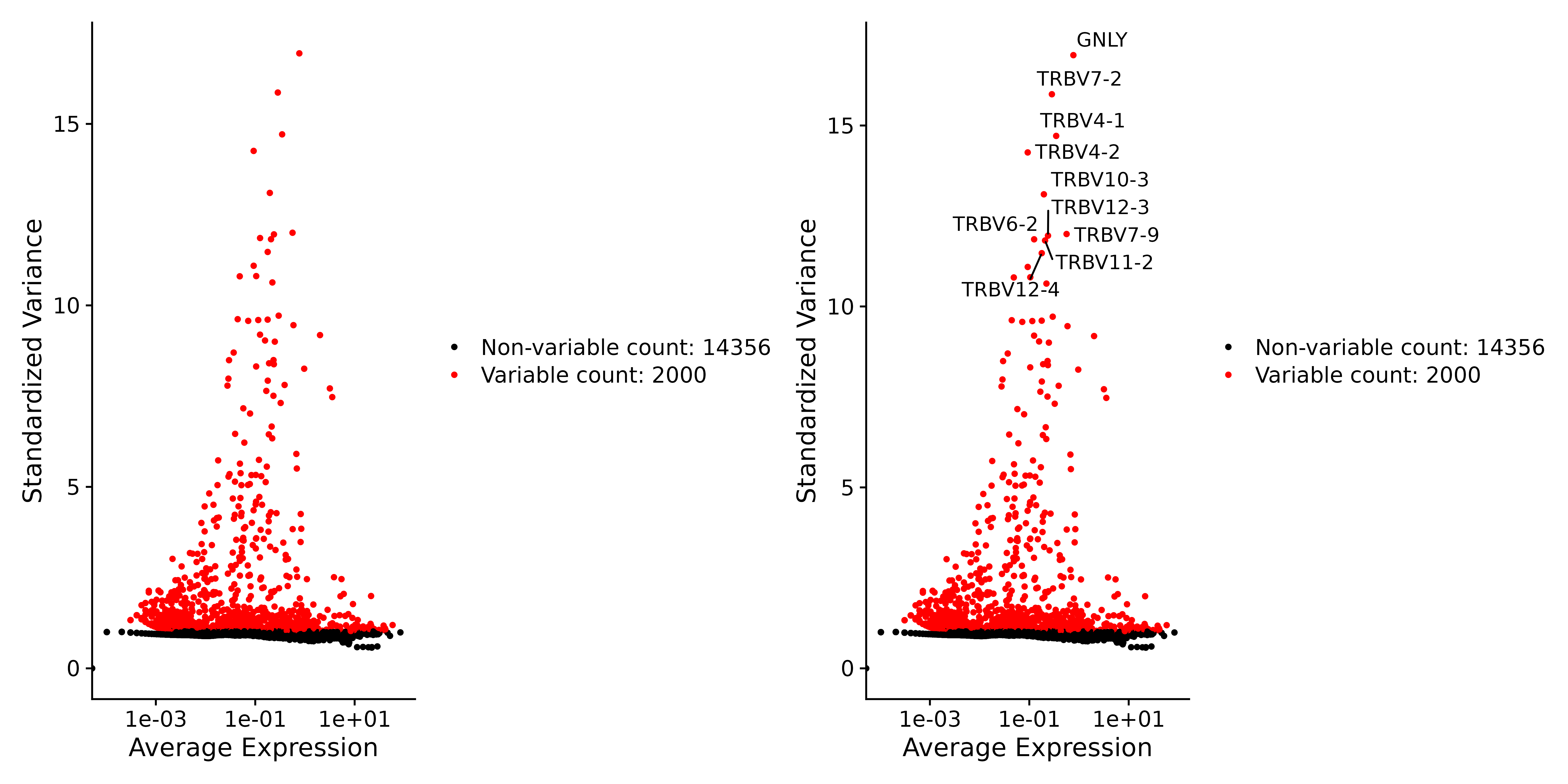
5. 데이터 스케일링 및 PCA 실행
단일 세포 전사체 데이터 분석에서 데이터 스케일링과 PCA는 데이터의 차원을 효과적으로 줄이고, 세포 간 변이의 주요 원인을 추출하며, 세포 이질성과 기능을 이해하는 데 중요한 정보를 제공할 수 있습니다.
1. 데이터 스케일링: 데이터 스케일링은 유전자 발현 데이터를 표준 정규 분포로 변환하는 것을 목표로 하는 선형 변환입니다. 즉, 데이터의 평균은 0이고 표준 편차는 1입니다. 이 변환은 서로 다른 유전자의 발현 수준 간의 차이를 줄이고 데이터를 더 정규화하는 데 도움이 됩니다. 스케일링을 통해 데이터에서 극단적인 값이나 이상치가 미치는 영향을 줄일 수 있고, 데이터 분포를 더 균등하게 만들 수 있으며, 후속 분석 단계에 더 안정적인 입력을 제공할 수 있습니다. 이 단계는 PCA(주성분 분석)를 위한 준비이기도 합니다. PCA는 표준화된 데이터에서 더 나은 성능을 보이기 때문입니다.
2. PCA 실행:
- PCA는 직교 축을 변환하여 데이터를 새로운 좌표계로 변환하는 통계적 방법으로, 데이터의 모든 투영에서 첫 번째로 큰 분산은 첫 번째 좌표(첫 번째 주성분이라고 함)에 있고, 두 번째로 큰 분산은 두 번째 좌표에 있는 식으로 계속됩니다.
- 단일 세포 전사체 분석에서 PCA는 유전자 발현의 차이에 따라 개별 세포를 분리하는 데 사용됩니다. 각 셀은 고차원 공간의 점으로 볼 수 있으며, PCA는 가능한 한 많은 변동 정보를 유지하면서 이러한 점을 저차원 공간에 다시 표현합니다.
- 각 주성분(PCA)은 본질적으로 세포의 "메타 특징", 즉 세포 변화의 주요 방향을 나타냅니다. PCA 결과에서 주성분이 더 전방에 있을수록 데이터 변화에서 가중치가 커지며, 이는 더 많은 정보를 포함하고 있음을 의미합니다.
- PCA를 통해 세포의 고차원 발현 정보를 여러 가지 주요 주성분으로 압축할 수 있으며, 이를 후속 클러스터링 분석, 시각화 또는 기타 다운스트림 분석에 사용할 수 있습니다.
all.genes <- rownames(seurat.subset)
seurat.subset <- ScaleData(seurat.subset, features = all.genes)
seurat.subset <- RunPCA(seurat.subset, features = VariableFeatures(object = seurat))##### Examine and visualize PCA results a few different ways-------------
print(seurat.subset[["pca"]], dims = 1:5, nfeatures = 5)
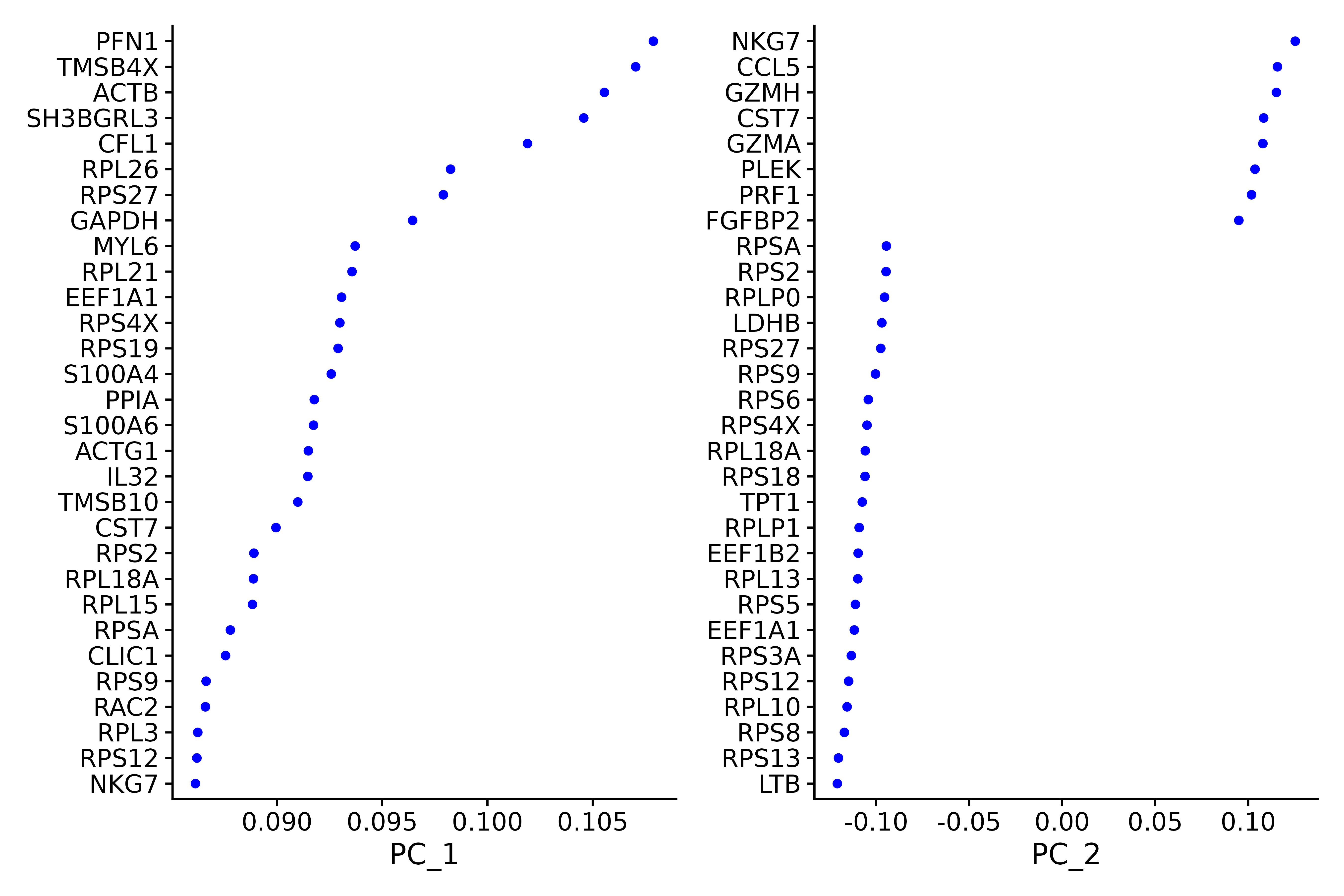
p1 <- VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
p1
ggsave("VizDimLoadings_5.png", p1, width = 9, height = 6, dpi=600)
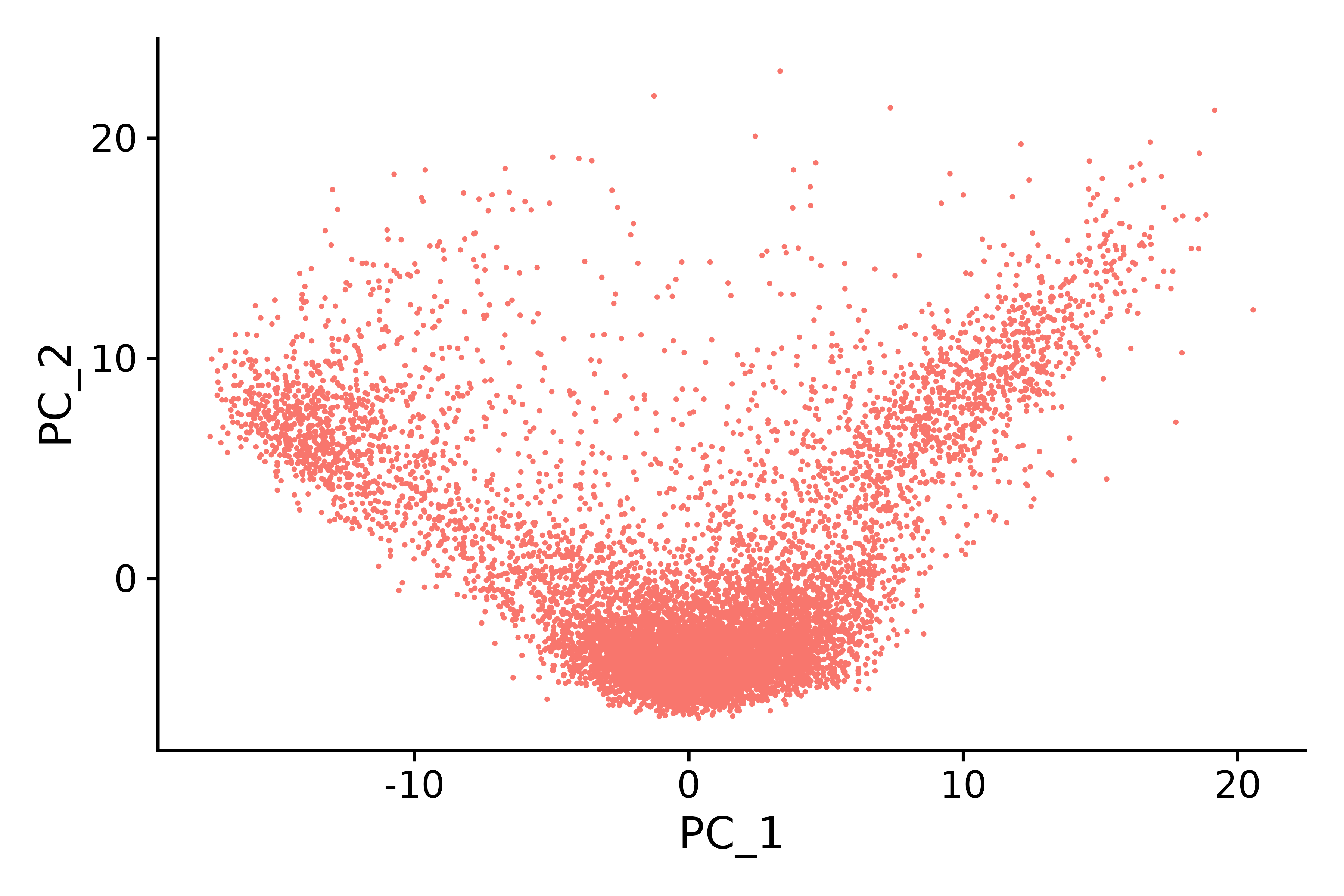
p2 <- DimPlot(seurat.subset, reduction = "pca") + NoLegend()
p2
ggsave("pca_6.png", p2, width = 6, height = 4, dpi=600)
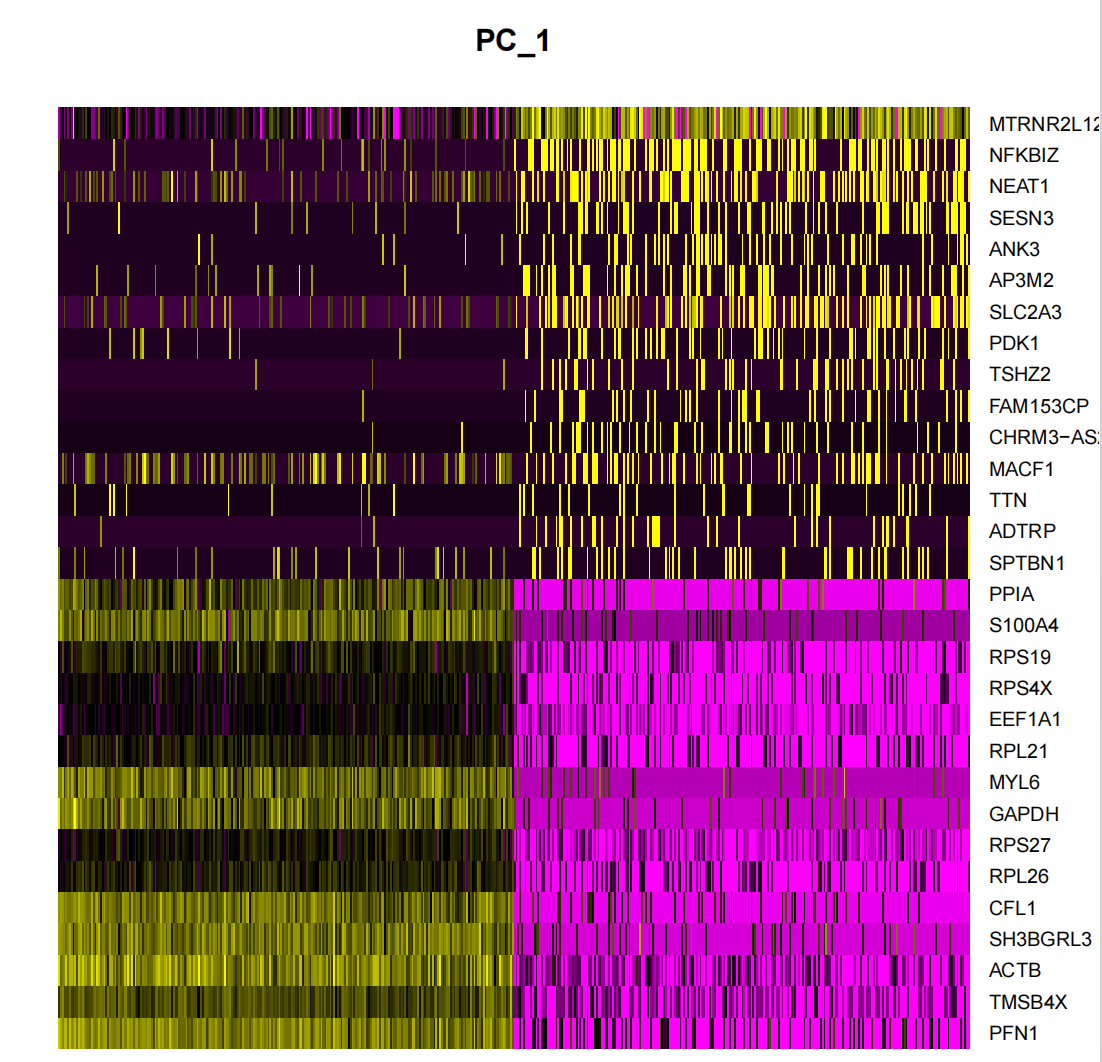
p3 <- DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
p3
ggsave("pca_1dims.png", p3, width = 6, height = 4, dpi=600)
p4 <- DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
p4
ggsave("pca_15dims.png", p4, width = 10, height = 10, dpi=600)
p5 <- ElbowPlot(seurat.subset)
p5
ggsave("ElbowPlot.png", p5, width = 10, height = 10, dpi=600)
DimHeatmap()은 연구자들이 데이터 세트의 주요 이질성 원인을 쉽게 식별하고 탐색할 수 있는 강력한 데이터 분석 도구입니다. 주성분 분석(PCA)을 수행한 후, DimHeatmap()은 후속 심층 분석에 포함해야 할 주성분(PC)을 결정하는 데 특히 유용합니다. 이 프로세스에는 PCA 점수에 따라 셀과 기능의 순위를 매기는 작업이 포함되어 분석의 정확성과 관련성을 보장합니다.
또한, DimHeatmap()을 사용하면 사용자는 데이터 스펙트럼의 양쪽 끝에서 극단적인 특성을 나타내는 "극단적인" 셀을 구체적으로 표시하기 위해 특정 개수의 셀을 설정할 수 있습니다. 이 접근 방식은 데이터의 가장 중요한 부분에 초점을 맞추고 전체 시각화 프로세스를 가속화하기 때문에 대용량 데이터 세트로 작업할 때 플로팅 효율성을 크게 향상시킵니다.

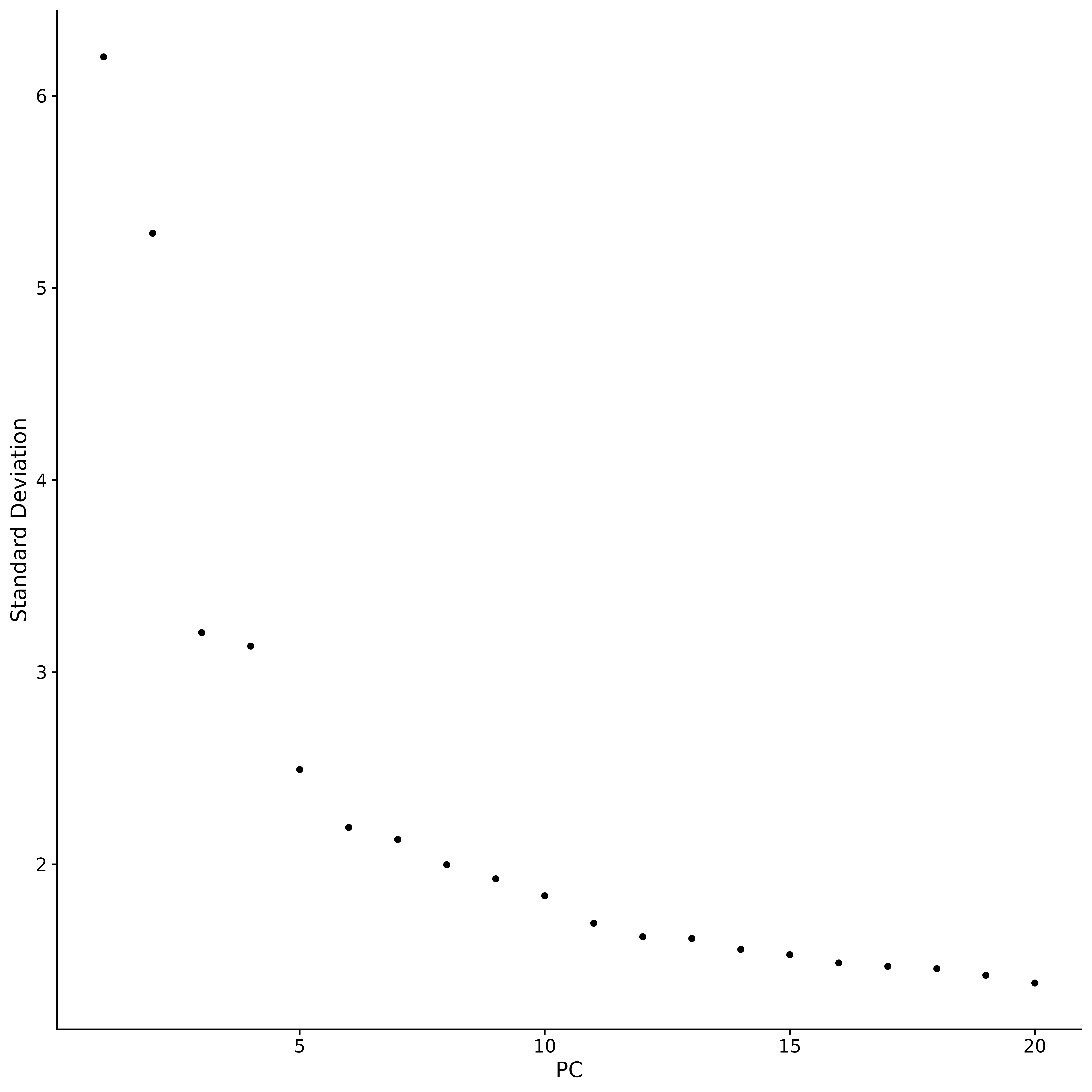
주성분 분석(PCA)에서는 후속 분석을 위해 얼마나 많은 주성분을 보관해야 하는지 결정하기 위해 "엘보 플롯"을 사용하는 것이 일반적인 관행입니다. 이 그래프는 대부분의 실제 신호가 처음 17개 주성분에 포착되었음을 보여줍니다. 엘보 플롯에서 우리는 전환점인 "엘보"를 찾습니다. 이 지점 이전에는 주성분을 추가할 때마다 데이터의 설명력이 크게 향상되지만, 이 지점 이후에는 주성분을 추가하여 얻는 이점이 줄어들기 시작합니다.이는 평형 상태에 도달했음을 나타냅니다.. 엘보우 플롯의 분석 결과를 토대로 후속 분석을 위해 첫 번째 17개 주성분을 선택했습니다. 주성분을 더 추가해도 데이터 판별력이 크게 향상되지 않고, 오히려 불필요하게 계산량이 늘어나기 때문입니다. 주성분의 개수를 계속 늘리면 데이터의 판별력이 약간 향상될 수 있지만, 그 향상 정도는 크지 않고 오히려 계산 비용이 크게 증가하게 됩니다. 따라서 17개의 주성분을 선택하는 것은 판별력과 계산 효율성의 균형을 맞추는 합리적인 선택입니다.
pdf("dimreduction_plots.pdf")#保存为 pdf 文件
VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
DimPlot(seurat.subset, reduction = "pca")
DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
ElbowPlot(seurat.subset)
dev.off()seurat.subset.dimReduction <- seurat.subset
seurat.subset.dimReduction <- FindNeighbors(seurat.subset.dimReduction, reduction="pca", dims = 1:17)
saveRDS(seurat.subset.dimReduction, file = "~/home/output/data/seurat_pre_FindClusters.rds")
# seurat.subset.dimReduction <- readRDS("~/home/output/data/seurat_pre_FindClusters.rds")6. 세포 클러스터링
### 3. Find clusters,聚类
```{r find_clusters}
seurat.final <- FindClusters(object = seurat.subset.dimReduction, resolution = 0.1)
head(Idents(seurat.final), 5)
seurat.final <- RunUMAP(seurat.final, reduction="pca", dims=1:17)
#Visualize with UMAP
p1<- DimPlot(seurat.final, reduction = "umap")
p1
ggsave("find_clusters.png", p1, width = 6, height =5 , dpi=600)
saveRDS(seurat.final, file = "~/home/output/data/seurat_final.rds")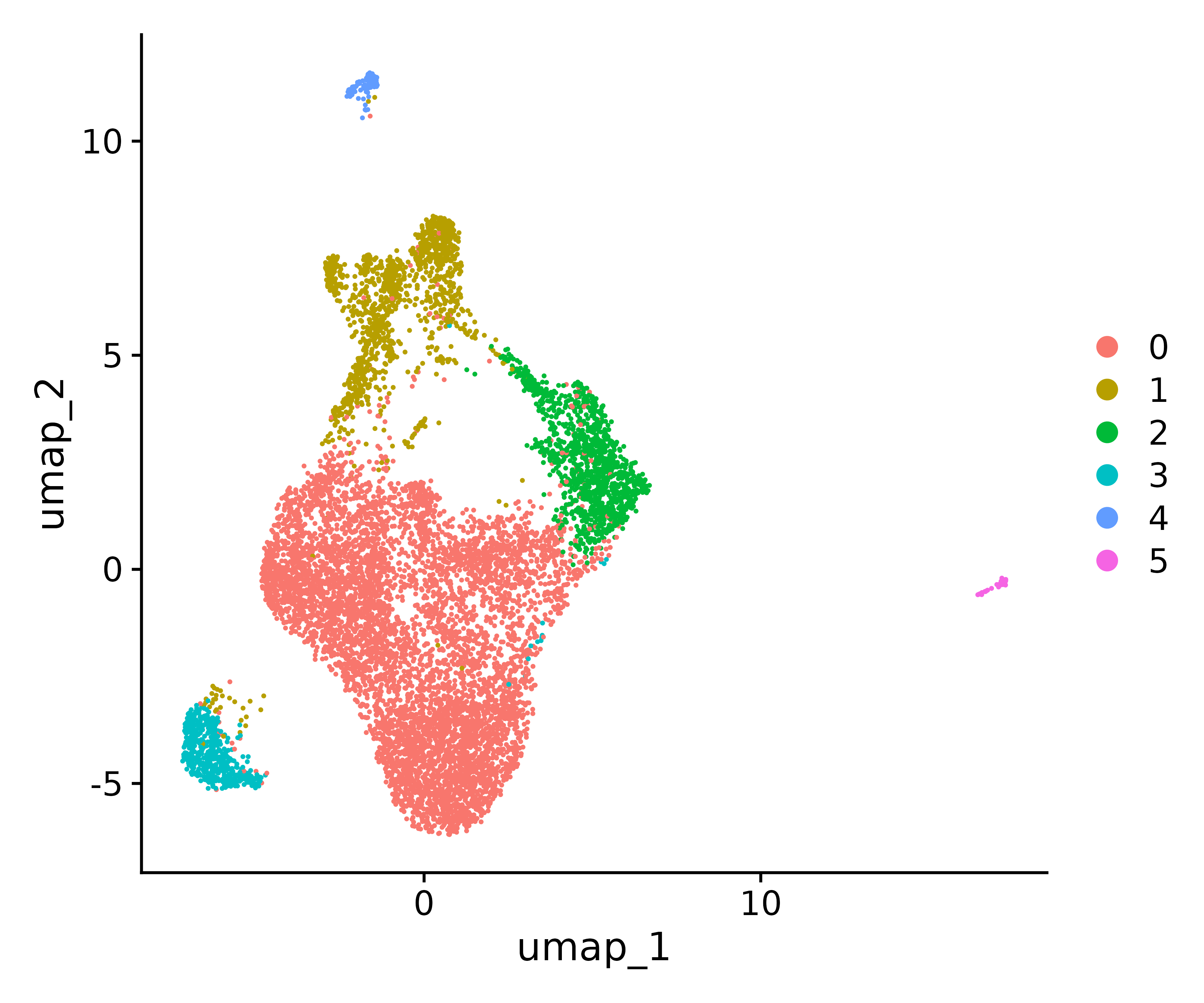
심상
다양한 클러스터에서 유전자 발현을 보기 위한 세 가지 일반적인 플롯은 다음과 같습니다.
1. 히트맵 2. 버블맵 3. 피처플롯
table(Idents(seurat.final))
# find markers between cluster1 and cluster 2
##用来找 cluster 之间的寻找差异表达特征
cluster2.markers <- FindMarkers(seurat.final, ident.1 = 1,ident.2 = 2)
head(cluster2.markers, n = 5)
# find markers for every cluster compared to all remaining cells, report only the positive ones
#找每个 cluster 的 marker 基因
pbmc.markers <- FindAllMarkers(seurat.final, only.pos = TRUE)
ClusterMarker_noRibo <- pbmc.markers[!grepl("^RP[SL]", pbmc.markers$gene, ignore.case = F),]
ClusterMarker_noRibo_noMito <- ClusterMarker_noRibo[!grepl("^MT-", ClusterMarker_noRibo$gene, ignore.case = F),]
###去除核糖体基因和线粒体基因的影响
ClusterMarker_noRibo_noMito %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
###展示每个 cluster 的 top5 的基因
heatmap <- DoHeatmap(seurat.final, features = top5$gene) + NoLegend()
ggsave("heatmap.png", heatmap, width = 10, height =5 , dpi=600) 이 그림은 마커 유전자를 보여주는 열 지도입니다.
이 그림은 마커 유전자를 보여주는 열 지도입니다.
g =unique(top5$gene)
##定义 top5 的 gene 列,作为基因集 g,也可以自定义
p <- DotPlot(seurat.final,features=g)+
coord_flip() + # 翻转坐标轴
theme(
panel.grid = element_blank(), # 移除网格线
axis.text.x = element_text(angle = 0, hjust = 0.5, vjust = 0.5) # 旋转 x 轴标签
)
ggsave("allmarker_dotpot.png", p, width = 8, height =7 , dpi=600)
#气泡图展示 marker 基因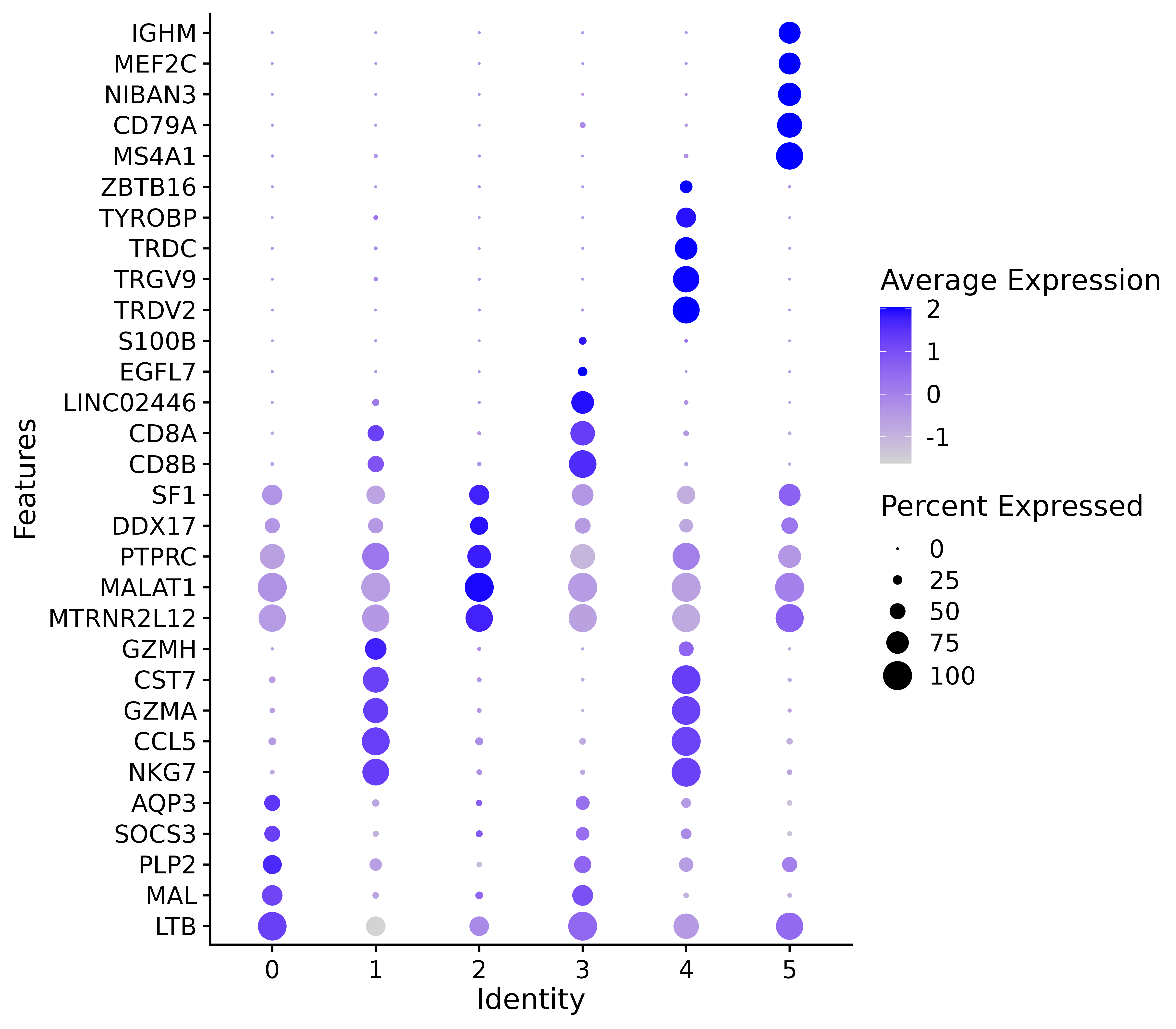
이 그림은 마커 유전자를 보여주는 거품형 차트입니다.
# 加载 dplyr 包
library(dplyr)
# 提取 cluster 和 gene 列
cluster_gene_mapping <- top5 %>%
select(cluster, gene)
# 查看结果
head(cluster_gene_mapping,30)
write.csv(cluster_gene_mapping,"cluster_allmarkers_TOP5.csv", row.names = p <- FeaturePlot(seurat.final, features = g)
ggsave("allmarker_featurepot.png", p, width = 15, height =15 , dpi=600)
###查看自定义感兴趣的基因
p2 <- FeaturePlot(seurat.final, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
ggsave("allmarker_featurepot2.png", p2, width = 15, height =15 , dpi=600)


7. 데이터 분석
이 단계에서는 단일 세포 전사체 데이터 분석에서 특정 마커 유전자를 사용하여 세포 유형을 결정하고 정의하는 방법을 설명합니다.
상위 마커 유전자 등을 기반으로 세포 유형을 결정했다고 가정하면 정체성 유형을 정의할 수 있습니다.
0 1 2 3 4 5 “CD4 T”, “NK”, “Treg”, “CD8 T”, “γδ T 세포”, “B 세포”
(다음 댓글은 참고용일 뿐 실제 댓글이 아닙니다.)
new.cluster.ids <- c("CD4 T", "NK", "Treg", "CD8 T", "γδ T cell",
"B cell")
names(new.cluster.ids) <- levels(seurat.final)
seurat.final <- RenameIdents(seurat.final, new.cluster.ids)
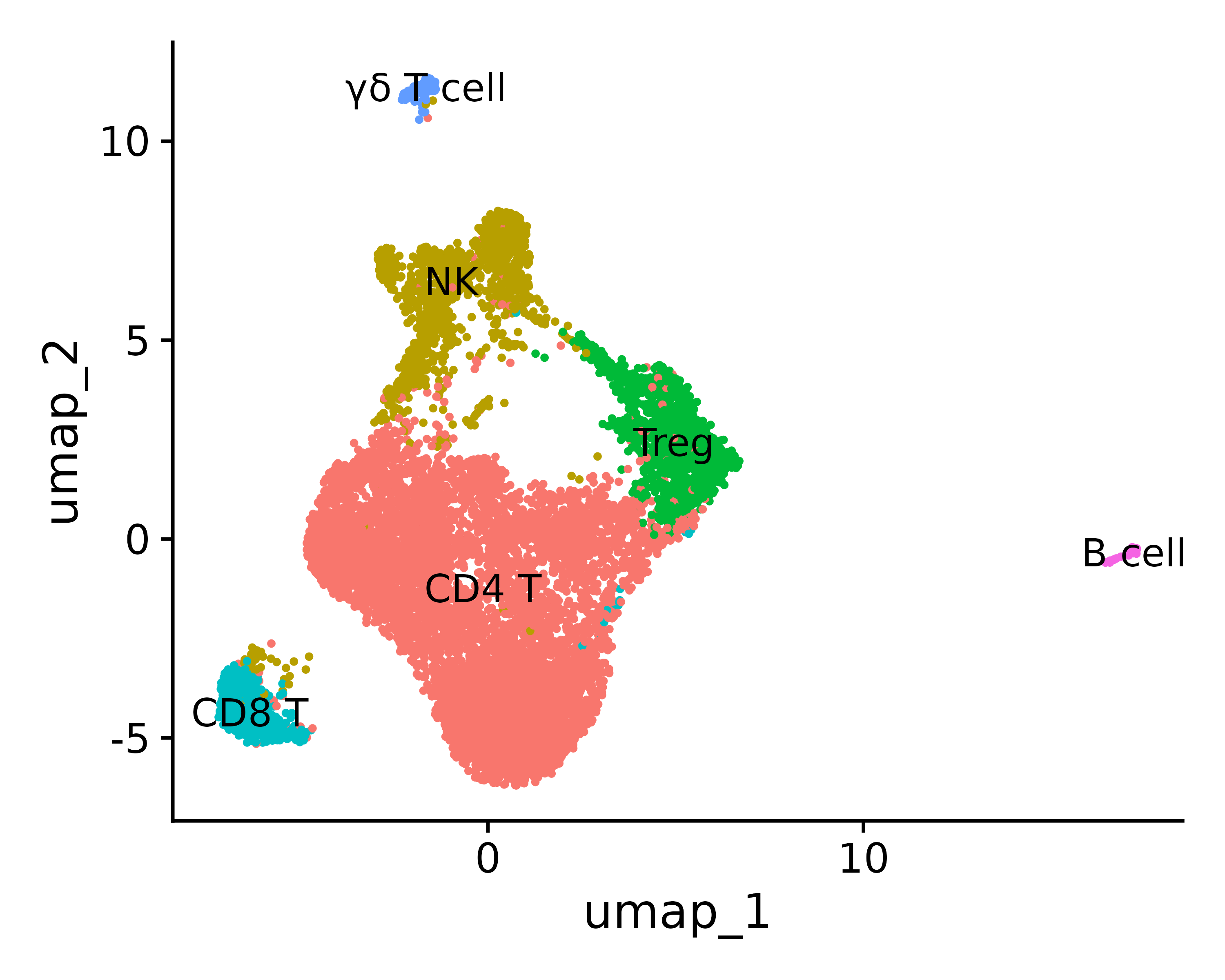
p <- DimPlot(seurat.final, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
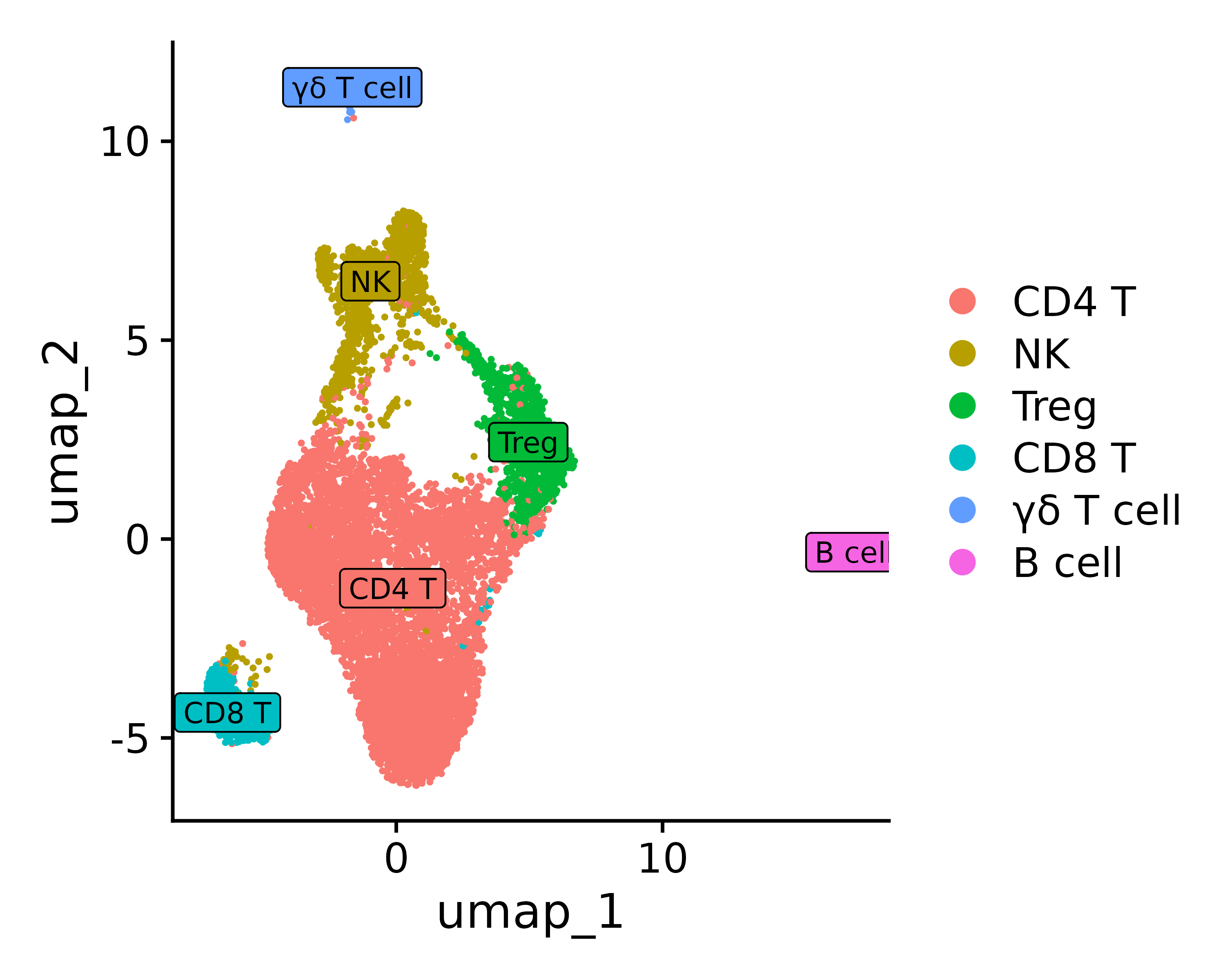
p3 = DimPlot(seurat.final, reduction = "umap", label = TRUE, label.size = 3, pt.size = .3, label.box = T)
ggsave("annotation_umap.png", p, width = 5, height =4 , dpi=600)
ggsave("annotation_umap2.png", p3, width = 5, height =4 , dpi=600)