Command Palette
Search for a command to run...
단백질 생성/접힘/역접힘을 지원하기 위해 후난대학교/중국과학원/ByteDance는 모든 원자 설계 및 기능 최적화를 달성하기 위해 APM 모델을 제안했습니다.
생명 활동의 주요 실행자인 단백질은 종종 다중 사슬 복합체의 형태로 기능을 수행합니다. 항체-항원 인식부터 효소-기질 결합에 이르기까지 다중 사슬 단백질 간의 정확한 상호작용은 생명 메커니즘을 이해하는 데 핵심적인 역할을 합니다.그러나 현재 AI 기반 단백질 모델링 분야는 상당한 "단일 사슬 편향"을 보입니다. AlphaFold 및 ESM 시리즈와 같은 모델이 단일 사슬 단백질의 접힘 및 설계에서 획기적인 진전을 이루었지만, 다중 사슬 복합체 모델링은 아직 초기 단계에 있습니다.
다중 사슬 단백질을 처리하는 기존 방법은 일반적으로 "가짜 서열 연결" 전략을 채택하여 다중 사슬을 단일 사슬로 처리합니다.이 방법은 사슬 간 상호작용의 자연적 발현을 심각하게 제한합니다. 실제 생물학적 복합체에서 사슬 간 공간적 위치와 결합 계면(수소 결합 및 소수성 상호작용 등) 사이의 원자 수준 상호작용은 선형 연결을 통해 정확하게 모델링할 수 없습니다. 또한, 모든 원자 구조를 생성하는 것은 두 가지 과제에 직면합니다. 아미노산 측쇄의 복잡한 형태와 강한 서열-구조 의존성으로 인해 다중 사슬 복합체의 새로운 설계는 이 분야에서 어려운 과제입니다.
이러한 연구 격차를 메우기 위해 후난대학교, 중국과학원대학교, ByteDance Seed 팀은 다중 사슬 단백질 복합체에 맞게 특별히 설계된 전 원자 단백질 생성 모델인 APM(All-Atom Protein Generative Model)을 제안했습니다. APM은 모든 원자 구조를 갖는 다중 사슬 복합체를 직접 생성할 수 있을 뿐만 아니라 접힘 및 역접힘과 같은 기본 작업을 지원하고 항체 및 펩타이드와 같은 기능성 단백질의 설계에서 뛰어난 성능을 보여줍니다.
이 연구 결과는 "단백질 복합체 설계를 위한 전원자 생성 모델"이라는 제목으로 ICML 2025에 선정되었습니다.
연구 하이라이트:
* 다중 체인 네이티브 모델링: 가상 시퀀스 연결을 포기하고 다중 체인의 독립적인 공간 분포와 바인딩 인터페이스 간의 원자 수준 상호 작용을 직접 학습합니다.
* 전체 원자 표현 최적화: 계산 효율성과 구조적 세부 사항의 균형을 맞추고 아미노산 유형, 백본 프레임워크 및 측쇄 비틀림 각도의 공동 표현을 통해 원자 수준의 구조 생성을 달성합니다.
* 시퀀스-구조 의존성 강화: 노이즈 프로세스와 양방향 작업 훈련(접기/펼치기)을 분리하여 시퀀스와 구조 간의 깊은 연관성을 유지합니다.

서류 주소:
공식 계정을 팔로우하고 "APM"으로 답글을 남겨 전체 PDF를 받아보세요.APM 단백질 생성 데이터 세트:
더 많은 AI 프런티어 논문:
데이터 세트: 단일 체인에서 다중 체인까지 풍부한 샘플
APM은 단일 사슬 및 다중 사슬 단백질의 구조와 서열 정보를 통합하는 신중하게 구성된 다중 소스 단백질 데이터 세트를 기반으로 학습되어 모델에 대한 풍부한 학습 자료를 제공합니다.
단일 사슬 데이터세트는 다중 소스 융합 및 고품질 필터링을 통해 사슬 내 모델링을 위한 풍부한 기반을 제공합니다. 총 187,494개의 샘플을 포함하고 있으며, 다양한 단백질 유형과 기능 범주를 포괄합니다. 데이터는 주로 다음 3개의 권위 있는 데이터베이스에서 제공됩니다.
* PDB 데이터베이스: MultiFlow 데이터 처리 과정 후 18,684개 샘플이 스크리닝되었습니다.
* Swiss-Prot 데이터베이스: pLDDT>85의 고품질 구조를 선택하여 140,769개 샘플을 얻었습니다.
* AFDB 데이터베이스: 더욱 엄격한 선별 기준을 사용하여 pLDDT>95인 샘플이 보존되어 총 28,041개의 샘플이 생성되었습니다.
다중 사슬 단백질 데이터세트는 2~6개 사슬로 구성된 단백질 복합체를 포함하는 총 11,620개의 샘플을 포함하고 있으며, 이는 다중 사슬 모델링에 핵심적인 데이터 지원을 제공합니다. 다중 사슬 단백질 데이터는 PDB(Biological Assemblies) 생물학적 어셈블리 데이터에서 가져왔습니다. 후속 작업에서 정보 유출을 방지하기 위해 연구팀은 세 가지 유형의 샘플을 제외했습니다. SAbDab 항체 데이터베이스에 존재하는 샘플, 길이가 30 미만인 사슬(펩타이드로 간주)을 포함하는 샘플, 길이가 2,048을 초과하거나 클러스터 ID가 없는 샘플입니다.
연구진은 모델의 일반화 능력을 개선하기 위해 훈련 과정에서 다중 사슬 샘플을 무작위로 다듬었습니다. 잔류물이 384개가 넘는 샘플의 경우, 사슬 간 결합 인터페이스의 잔류물 쌍을 중심으로 가장 가까운 384개 아미노산을 유지했습니다.이러한 가지치기 전략은 메모리 오버플로 문제를 피하면서 모델이 주요 바인딩 영역에 집중할 수 있도록 보장합니다.또한, 연구진은 단일 사슬 데이터와 다중 사슬 데이터를 비례적으로 혼합하여 단일 사슬 데이터의 풍부함을 활용하여 사슬 내 모델링 성능을 향상시켰습니다. 각 샘플링 위치에는 지리적 위치(사슬 간 상호작용 부위), 구조적 특성(예: 이차 구조 유형), 서열 특징(아미노산 유형 및 보존)을 포함한 풍부한 메타데이터가 첨부되어 있습니다. 이 정보는 모델이 서열, 구조, 기능 간의 매핑 관계를 학습하는 데 다차원적인 단서를 제공합니다.
APM 단백질 생성 데이터 세트:
모델 아키텍처: 3개 모듈로 구성된 협업적 전체 원자 생성 프레임워크
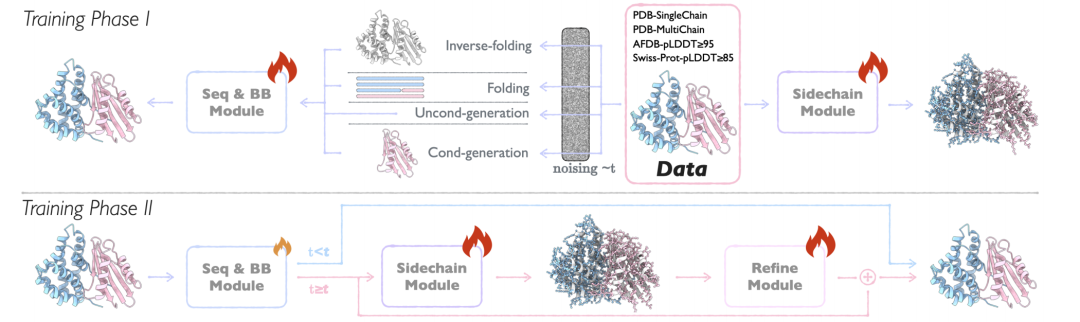
APM의 핵심 아키텍처는 명확한 기능을 갖춘 세 가지 모듈로 구성되어 있습니다. 시퀀스 및 백본 생성 모듈(Seq&BB 모듈), 사이드체인 생성 모듈(Sidechain 모듈), 정제 모듈입니다.혁신적인 설계를 통해 다중 사슬 단백질의 다양한 설계 과제를 지원하는 동시에 시퀀스에서 전체 원자 구조까지의 종단 간 생성이 달성되었습니다.
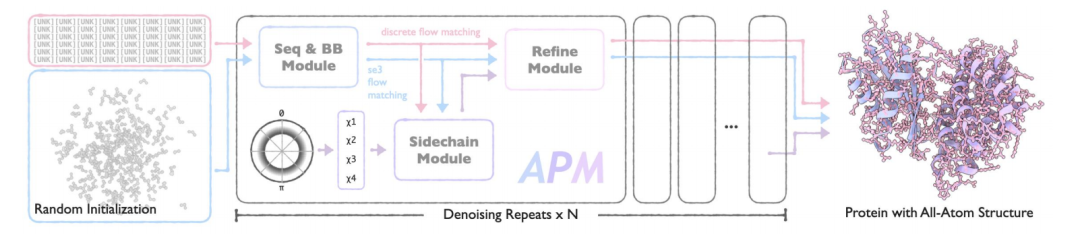
Seq&BB 모듈
이 모듈은 APM의 기반입니다. 흐름 매칭 방식을 채택하여 서열 및 단백질 백본의 공동 생성을 실현하고, 잔류물 수준에서 서열-구조 협업 모델링 작업을 처리할 수 있습니다.시퀀스와 구조의 노이즈 과정을 분리함으로써 시퀀스-구조 의존성 관계의 손상을 줄이고, 폴딩/역폴딩 작업을 50% 확률로 수행하여 양방향 의존성 학습을 강화합니다. 이 모듈의 핵심 혁신은 다음과 같습니다.
* 노이즈 분리 프로세스:시퀀스 노이즈 프로세스와 구조 노이즈 프로세스를 분리하면 기존 방식에서 발생하는 모달 간 종속성 손상 문제를 방지할 수 있습니다. 노이즈 시퀀스와 노이즈 백본은 서로 다른 시간 단계에서 독립적으로 샘플링되므로, 모델이 양방향 시퀀스-구조 종속성을 학습할 수 있습니다.
* SE(3) 흐름 매칭:단백질 백본의 공간적 변형 특성을 고려하여 이동 및 회전 부분을 별도로 처리하기 위해 3차원 특수 유클리드 군(SE(3)) 흐름 매칭이 도입되었습니다.
* 멀티태스킹 학습:또한 무조건 생성, 조건 생성, 폴딩 및 역폴딩 작업을 지원하고, 혼합 작업 학습을 통해 모델의 일반화 능력을 향상시킵니다. 손실 함수에는 생성된 궤적의 부드러움을 보장하기 위해 흐름 매칭 손실과 일관성 손실이 포함됩니다.
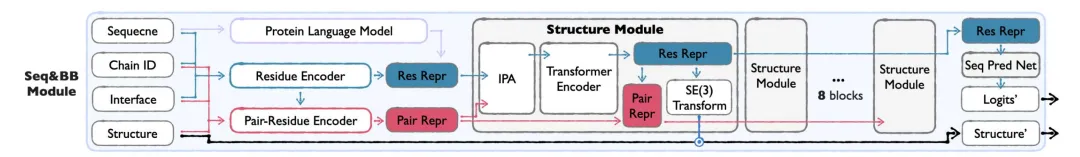
사이드체인 모듈
모든 원자 구조를 생성하기 위해 Sidechain Module은 Seq&BB가 생성한 시퀀스와 백본을 기반으로 아미노산 측쇄의 형태를 예측합니다.
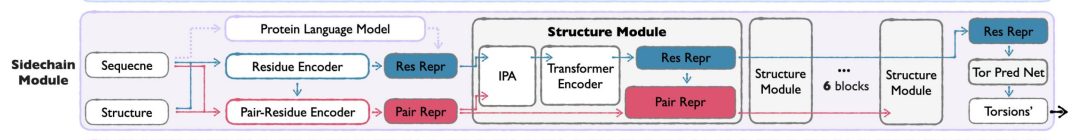
모듈은 다음과 같은 전략을 채택합니다.
* 비틀림 각도는 다음을 의미합니다.측사슬 구조는 측사슬 비틀림 각도(최대 4개의 회전 가능한 결합)로 매개변수화되어 계산 효율성과 원자 수준의 세부 사항 간의 균형을 이루며, 모든 원자 좌표의 직접 모델링의 복잡성을 피합니다.
* 2단계 훈련:첫 번째 단계는 측사슬 패킹 작업에 초점을 맞추고 실제 측사슬 형태의 분포를 학습합니다. 두 번째 단계에서는 예측된 구조로부터 실제 측사슬을 재구성하여 생성 시나리오에서 모델의 적용성을 보장합니다.
* 가벼운 디자인:Seq&BB 모듈과 비교했을 때, Sidechain 모듈은 구조적 블록을 덜 사용하고 숨겨진 차원도 더 작습니다.
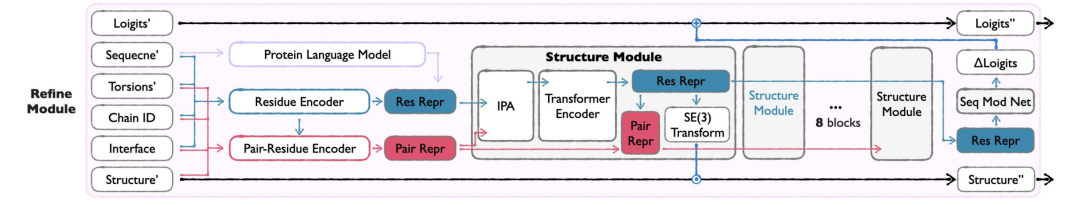
모듈 정제
APM의 마지막 링크인 Refine 모듈은 Seq&BB와 Sidechain 모듈의 출력을 통합하고, 손실을 보정하여 시퀀스와 백본을 최적화하고, 원자 충돌을 줄이며, 구조적 합리성을 향상시킵니다.전체 원자 정보는 서열 및 주쇄 구조를 최적화하고, 구조적 충돌을 해결하며, 생성된 결과를 자연 단백질에 더 가깝게 만드는 데 사용됩니다. 이 모듈은 입력 품질이 최적화를 지원하기에 충분한지 확인하기 위해 후기 생성 단계(t≥0.8)에만 활성화됩니다.
실험적 결론: APM의 획기적인 성능에 대한 다차원적 검증
APM의 실험적 검증은 단일 체인 기본 작업, 다중 체인 핵심 작업 및 다운스트림 기능 설계를 포괄하며 그 결과는 모두 우수합니다.
단일 사슬 단백질 작업: 전문가 모델과 비교 가능한 기본 기능
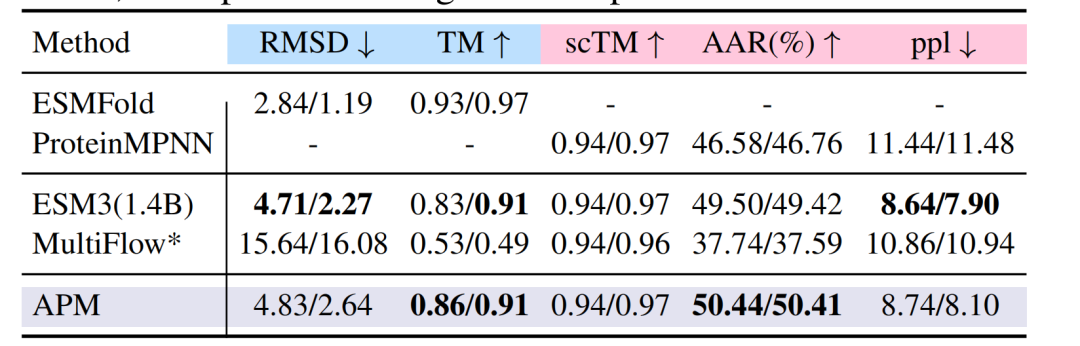
PDB 데이터셋에 대한 폴딩 작업에서APM의 RMSD는 4.83/2.64입니다.TM 점수는 0.86/0.91로 ESM3, MultiFlow 및 기타 모델의 성능과 유사했습니다. 역방향 폴딩 작업에서 아미노산 회수율(AAR)은 50.44%에 도달하여 ProteinMPNN의 46.58%를 넘어섰습니다.
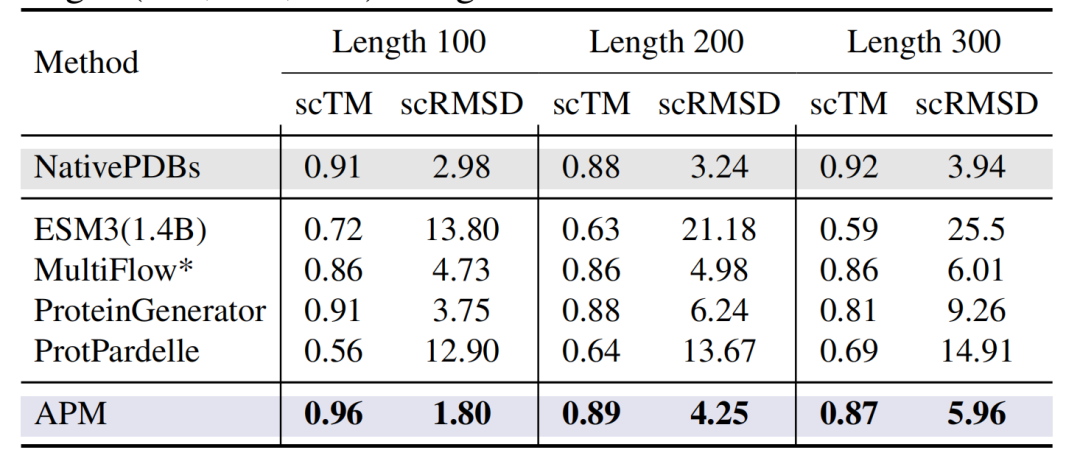
또한, 아래 그림과 같이 무조건 생성된 100~300잔기 길이의 단백질에서는APM의 scTM은 0.96(길이 100) 정도로 높고, scRMSD는 1.80 정도로 낮습니다.ESM3(1.4B) 및 ProtPardelle과 같은 모든 원자 설계 모델보다 상당히 우수합니다.
다중 사슬 단백질 작업: 네이티브 모델링의 핵심 장점
접힘과 펼침 실험에서,2-6 사슬 복합체에서 APM의 폴딩 성능은 12.6/13.67로 Boltz-1보다 낮지만, MSA를 적용하지 않은 Boltz-1보다는 상당히 우수합니다. 역폴딩 scTM은 0.85/0.95로 MSA를 적용한 Boltz-1과 유사하여 서열-구조 연관의 타당성을 입증합니다. 실험 결과는 아래 그림에 나와 있습니다.
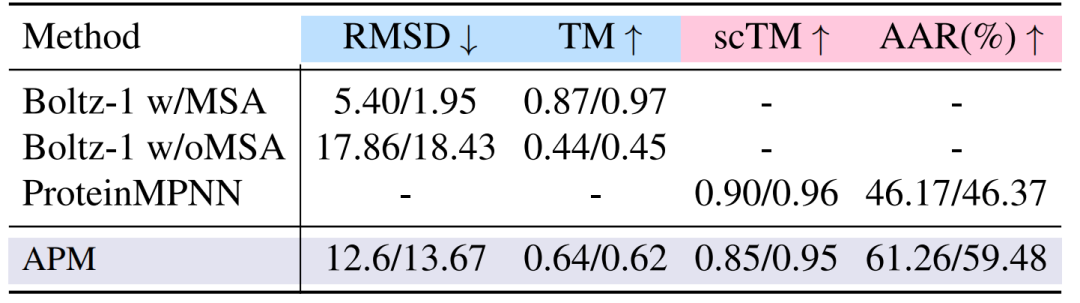
둘째,다중사슬 복합체는 강력한 결합 친화력을 가지고 있습니다.예를 들어 50-100의 사슬 길이를 취하면, 모든 원자가 이완된 후의 결합 에너지 ΔG_RAA는 -112.65/-116.98에 도달하는데, 이는 주 사슬만을 사용했을 때의 Chroma(-83.96/-86.66)와 APM_BB(-114.94/-114.45)보다 상당히 우수합니다. 이는 사슬 간 상호 작용을 모델링하기 위해서는 모든 원자 정보가 필요함을 증명합니다.
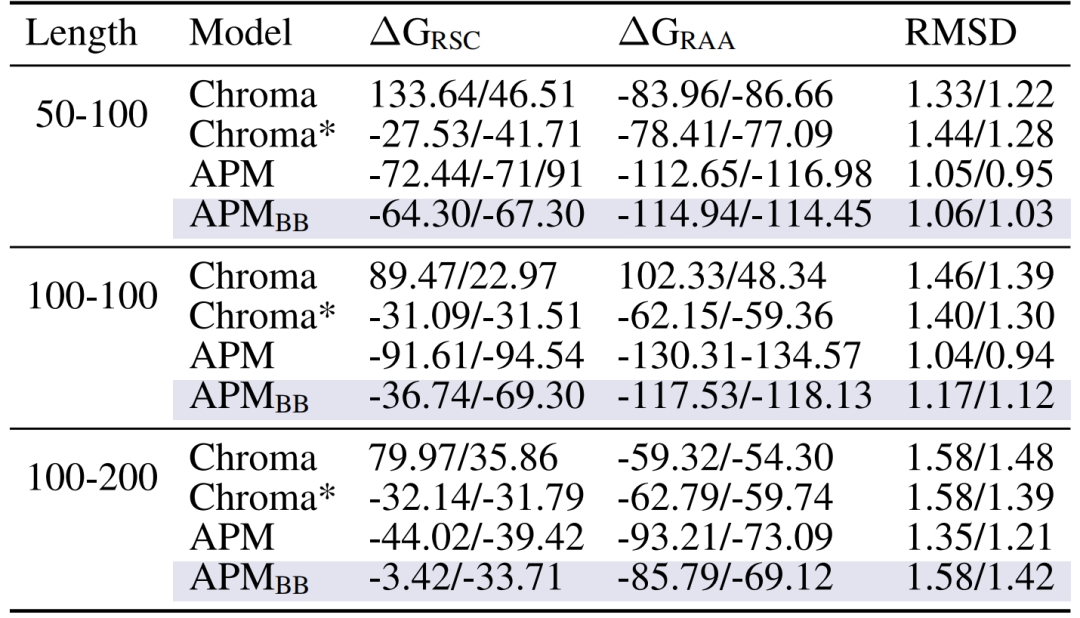
생성된 복합체 사이의 사슬 간 결합 친화력
하류 기능 설계: 항체 및 펩타이드 응용 분야의 획기적인 발전
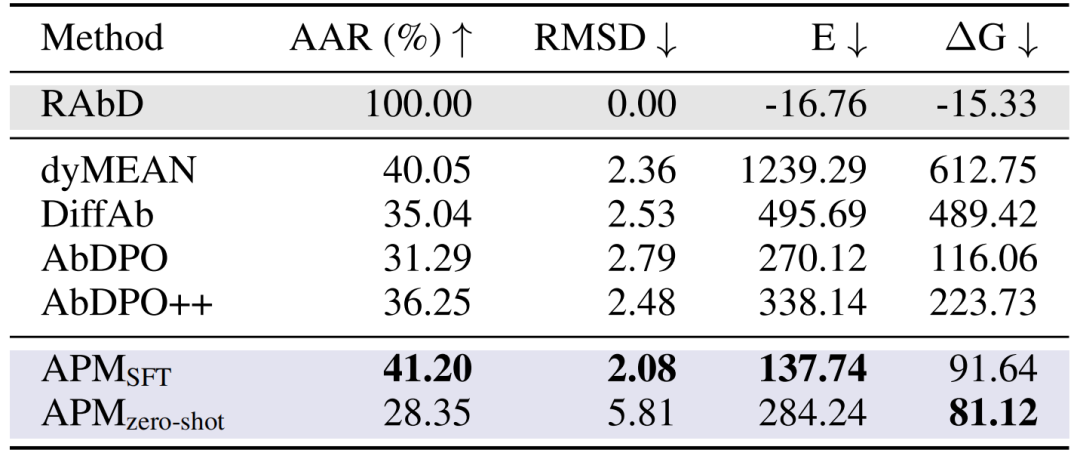
항체 CDR-H3 디자인:RAbD 벤치마크 테스트에서 APM의 AAR은 41.20%, RMSD는 2.08, 결합 에너지 ΔG는 91.64로 dyMEAN 및 DiffAb와 같은 방법을 능가했습니다. 무표본 항체로 생성된 항체의 서열은 천연 항체와 매우 다르지만, 결합 에너지는 더 우수하여(ΔG 81.12), APM의 보편적인 결합 능력을 입증했습니다.
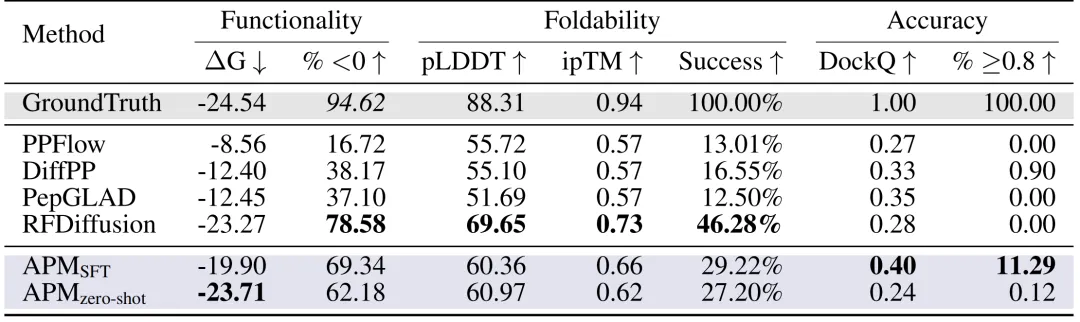
펩타이드 디자인:연구진은 PepBench와 LNR 데이터셋을 이용하여 기능성, 접힘성, 정확도라는 세 가지 핵심 측면에서 펩타이드 설계 방법을 종합적으로 평가했습니다. 아래 그림에서 볼 수 있듯이, APM(SFT)의 결합 에너지 ΔG는 -19.90에 도달했고, 69.341개의 TP3T 샘플에서 ΔG < 0이었으며, DockQ ≥ 0.8의 비율은 11.29%에 도달하여 PPFlow, PepGLAD 및 기타 방법을 크게 상회했습니다. 또한 접힘 안정성(pLDDT 60.36, ipTM 0.66)도 매우 우수했습니다.
산업-R&D 협업으로 전원자 단백질 생성 기술의 획기적인 발전이 이루어짐
모든 원자 단백질 생성이라는 최첨단 생물학 분야에서 학계와 기업은 끊임없이 연구를 거듭해 왔으며, 획기적인 결과들이 잇따라 발표되어 주목을 받고 있습니다.
학계에서는 DeepMind 팀이 출시한 AlphaFold3가 다중 스케일 구조 정보와 진화적 서열 데이터를 통합하여 전 원자 단백질 생성 분야에서 강력한 역량을 입증했습니다.복잡한 단백질 접힘 패턴의 정확한 모델링을 달성했습니다.특히 보조 인자와 금속 이온을 포함하는 전원자 복합체를 생성하는 작업에서 구조적 정확도와 에너지 합리성이 기존 방식에 비해 크게 향상되었습니다. 스탠퍼드 대학교 연구팀이 개발한 ESM-IF1은 다른 접근 방식을 취합니다. 방대한 진화적 서열 데이터로 학습된 암묵적 접힘 모델을 기반으로, 자연적인 구조적 특성을 가진 전원자 단백질 구조를 직접 생성할 수 있으며, 효소 활성 중심의 정밀한 구축에도 탁월한 성능을 보입니다.
업계 또한 이 분야에 적극적으로 진출하여 기술 혁신을 통해 산업 응용 분야를 확대하고 있습니다. 베이징 바이오지오메트리 바이오테크놀로지(Beijing Bio-Geometry Biotechnology Co., Ltd.)는 세계 최초의 풀 시나리오 원자 수준 단백질 모델인 GeoFlow V2를 출시했습니다. 이 모델은 단백질 원자의 정밀한 조절을 달성할 수 있는 엔드투엔드 확산 생성 프레임워크를 구축했습니다. 항체 CDR 영역의 풀 원자 설계에서,이를 통해 친화성과 안정성을 동시에 최적화하여 약물 개발 효율성을 크게 향상시킬 수 있습니다.미국 생명공학 기업인 인실리코 메디슨(Insilico Medicine)은 약물 표적 단백질 설계에 중점을 둔 단백질 생성 시스템을 개발했습니다. 이 시스템이 채택한 다중 제약 조건 생성 전략은 모든 원자 구조의 합리성을 보장하는 동시에 단백질과 저분자 약물 간의 결합 부위를 방향성 있게 최적화하여 후보 약물의 효율적인 스크리닝을 위한 탄탄한 기반을 제공합니다.
학계의 이러한 이론적 혁신과 비즈니스 커뮤니티의 응용 혁신은우리는 함께 실험실에서 산업 현장으로 전 원자 단백질 생성 기술을 확대하여 정밀 약물 개발, 새로운 생체 촉매 설계, 합성 생물학 분야의 획기적인 발전을 위한 핵심 지원을 제공하고, 앞으로 질병 치료와 생물 제조 분야에서 막대한 가치를 창출하기를 바랍니다.
참조 링크:
1.https://mp.weixin.qq.com/s/a0bl9ek90t_-y8wy69Yu6Q
2.https://mp.weixin.qq.com/s/P-5o-R1qZY52Pq1yK5j6cQ