LeCun은 UC Berkeley 등을 통해 전달되었습니다. 단백질 서열과 전체 원자 구조를 동시에 생성하는 다중 모드 단백질 생성 방법 PLAID를 제안했습니다.

지난 몇 년 동안 과학자들은 "생명의 암호"를 더 잘 풀기 위해 단백질의 구조와 구성을 계속 탐구해 왔습니다.단백질의 기능은 측쇄와 주쇄 원자의 정체성과 위치, 그리고 생물학적 특성을 포함한 구조에 의해 결정되는데, 이를 통칭하여 전체 원자 구조라고 합니다.그러나 측쇄 원자를 어디에 배치해야 할지 결정하려면 먼저 순서를 알아야 합니다. 따라서 모든 원자 구조 생성은 시퀀스와 구조를 동시에 생성해야 하는 다중 모드 문제로 볼 수 있습니다.
그러나 기존의 단백질 구조 및 서열 생성 방법은 일반적으로 서열과 구조를 독립적인 모드로 처리합니다. 구조 생성 방법은 일반적으로 주쇄 원자만 생성합니다. 전체 원자 설계를 목표로 하는 방법은 일반적으로 구조 예측과 안티 폴딩 단계 등을 번갈아가며 수행하기 위해 외부 모델을 사용해야 합니다.
이러한 과제를 해결하기 위해 캘리포니아 대학교 버클리(UC 버클리), 마이크로소프트 연구소, 제넨텍의 연구팀은 PLAID(Protein Latent Induced Diffusion)라는 다중 모드 단백질 생성 방법을 제안했습니다. 이 방법은 풍부한 데이터 모달리티(예: 시퀀스)에서 부족한 모달리티(예: 결정 구조)로 매핑하여 다중 모드 생성을 달성할 수 있습니다.연구진은 이 접근 방식을 검증하기 위해 유전자 온톨로지의 2,219개 기능과 생명의 나무 전체의 3,617개 유기체를 대상으로 실험을 수행했습니다.훈련 중에 구조적 입력을 사용하지 않더라도 생성된 샘플은 강력한 구조적 품질과 일관성을 보여줍니다.
관련 연구의 제목은 "시퀀스 전용 훈련 데이터에서 모든 원자 단백질 구조 생성"이며, 최고의 컨퍼런스인 ICLR 2025에 제출되었습니다. "AI 대부" 양리쿤도 이 업적을 소셜 플랫폼에 다시 게시했습니다.
PLAID 프로젝트 오픈소스 주소:
http://github.com/amyxlu/plaid
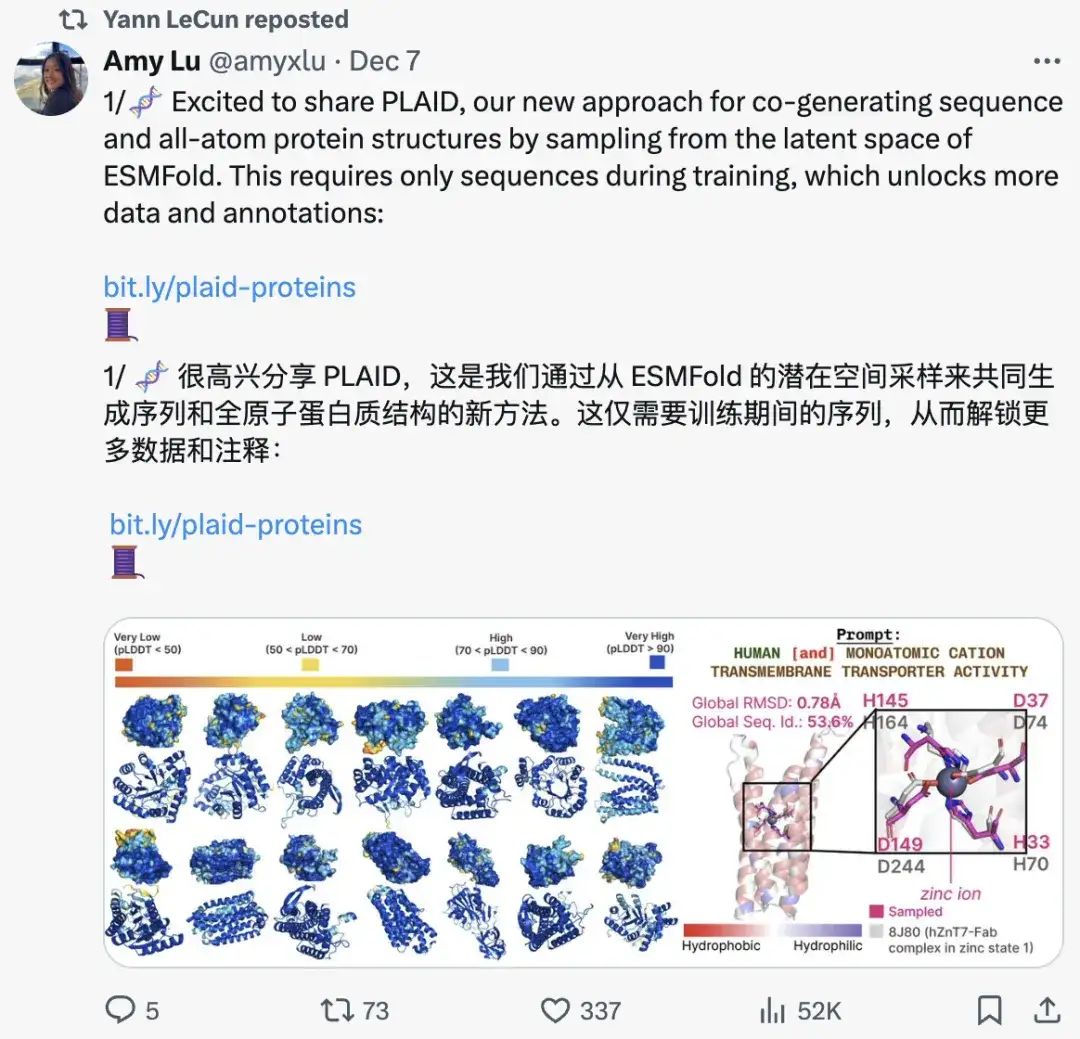
연구 하이라이트:
* 연구진은 대규모 단백질 언어 모델 ESMFold와 모든 원자 구조 생성에 초점을 맞춰, 훈련 중에 시퀀스 입력만 필요하고 시퀀스와 모든 원자 단백질 구조를 동시에 생성할 수 있는 제어 가능한 확산 모델을 제안했습니다.
* 이 접근 방식은 훈련 데이터가 아닌 사전 훈련된 가중치에 인코딩된 구조적 정보를 활용하고 제어 가능한 생성을 위한 시퀀스 주석의 가용성을 높입니다.
* 본 논문에서는 ESMFold 모델을 사용했지만, 이 방법은 모든 예측 모델에 적용될 수 있습니다.
서류 주소:
https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
오픈소스 프로젝트인 "awesome-ai4s"는 100개가 넘는 AI4S 논문 해석을 모아 방대한 데이터 세트와 도구를 제공합니다.
https://github.com/hyperai/awesome-ai4s
연구 하이라이트에 대한 간략한 개요
데이터세트
연구진은 57,595,205개의 시퀀스와 20,795개의 패밀리를 포함하는 2023년 9월에 출시된 Pfam 데이터베이스를 사용했습니다. PLAID는 UniRef나 BFD(약 20억 개의 시퀀스)와 같은 대규모 시퀀스 데이터베이스와 완벽하게 호환되지만, 이 연구에서는 Pfam을 사용하기로 했습니다. Pfam의 시퀀스 도메인에는 더 많은 구조적, 기능적 태그가 포함되어 있어 생성된 샘플에 대한 컴퓨터 시뮬레이션 평가가 더 편리하기 때문입니다. 또한 연구진은 검증을 위해 약 15%의 데이터를 보관했습니다.
Pfam 도메인이 유래된 생물체의 UniRef 코드는 Pfam FTP 서버에서 제공되는 Pfam-A.fasta 파일에서 사용할 수 있습니다. 연구진은 데이터 세트에 있는 모든 고유한 유기체를 분석하여 총 3,617개의 서로 다른 유기체를 찾아낸 다음, PLAID 방법의 효과를 검증하기 위해 이러한 유기체에 대한 실험을 수행했습니다.
모델 아키텍처
PLAID는 예측 모델의 잠재 공간에서 확산을 통한 다중 모드의 제어 가능한 단백질 생성을 위한 새로운 패러다임입니다.방법 개요는 아래 그림과 같습니다. 간단히 말해서, 4단계로 나뉩니다.
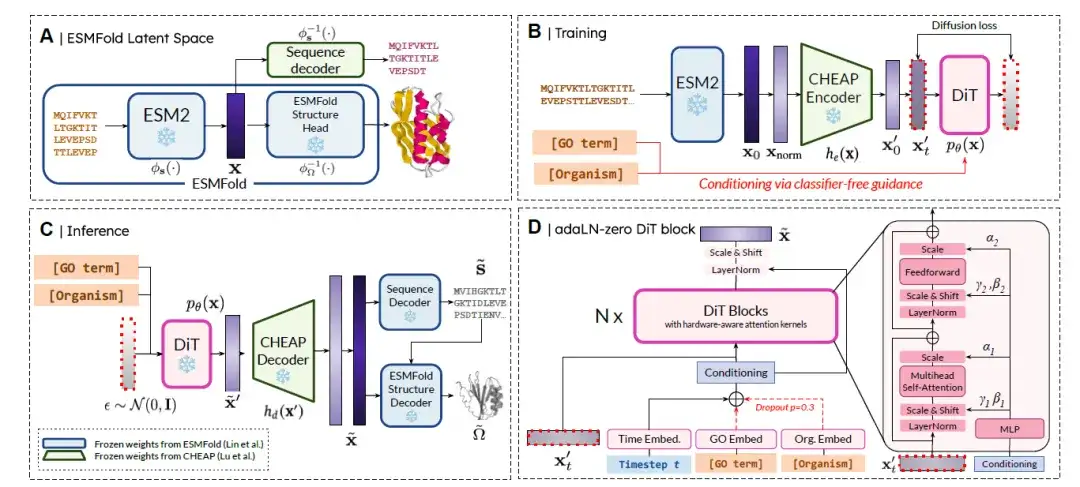
(A) ESMFold 잠재 공간:잠재 공간 p(x)는 시퀀스와 구조의 공동 임베딩을 나타냅니다.
(B) 잠재적 확산 훈련:목표는 확산 공식에 따라 pθ(x)에서 학습하고 샘플링하는 것입니다. 학습 효율성을 개선하기 위해 연구자들은 CHEAP 인코더 he(·)를 사용하여 압축 임베딩 x′ = he(x)를 얻어 확산 목적이 pθ(he(x))에서 샘플링이 되도록 했습니다.
(C) 추론:추론 시점에서 시퀀스와 구조를 모두 포착하기 위해 훈련된 모델을 사용하여 ˜x′ ∼ pθ(x′)를 샘플링한 다음 CHEAP 디코더를 사용하여 압축을 해제하여 ˜x = hd(˜x′)를 얻습니다. 임베딩은 CHEAP에서 훈련된 동결된 시퀀스 디코더에 의해 해당 아미노산 시퀀스로 디코딩됩니다. 잔류물 동일성 시퀀스와 ˜x는 ESMFold에서 학습된 동결 구조 디코더의 입력으로 사용되어 모든 원자 구조를 얻습니다.
(D) DiT 블록 아키텍처:연구진은 DiT(Diffused Transformer) 아키텍처와 adaLN-zero DiT 블록을 결합하여 조건부 정보를 융합했습니다. 기능적(즉, GO 용어) 및 생물체 클래스 레이블은 분류자 지침을 사용하지 않고 내장되었습니다.
연구 결과
연구진은 다양한 길이의 단백질에 대해 구조적 품질과 다양성 분석을 수행했으며, 그 결과는 아래 그림에 나와 있습니다.천연 단백질과 PLAID로 생성된 샘플은 길이가 달라도 일관된 측정 기준을 갖습니다.ProteinGenerator와 Protpardelle은 특정 길이에서 모드 붕괴를 보인 반면, Multiflow는 더 긴 시퀀스에서 다양성 감소를 보였습니다.
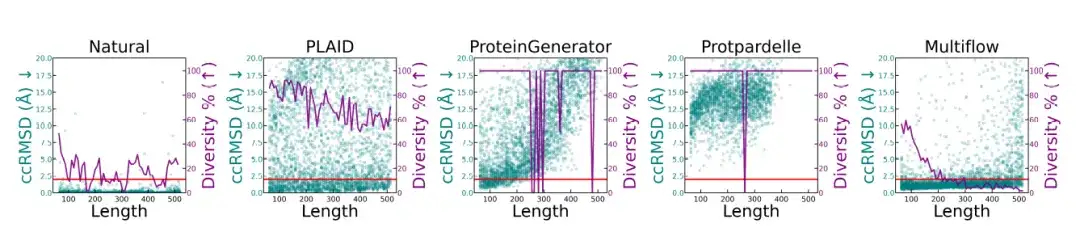
* 이 그림은 천연 단백질과 다양한 생성 방법을 비교하여 다양한 길이(잔기 64~512개)의 단백질의 구조적 품질(ccRMSD, 청록색 점)과 다양성(보라색 선, 전체 샘플에서 고유한 구조적 클러스터의 비율로 측정)을 보여줍니다. 빨간색 선은 2Å로 설계 임계값을 나타냅니다.
또한, 기준선 방식과 비교했을 때,PLAID가 생성하는 2차 구조의 다양성은 천연 단백질의 분포와 더욱 유사합니다.아래 그림에서 볼 수 있듯이 ProteinGenerator, Protpardelle 및 Multiflow는 2차 구조 분포에서 편차를 보이며, 기존의 단백질 구조 생성 모델은 일반적으로 β-시트 함량이 높은 샘플을 생성하는 데 어려움을 겪습니다.
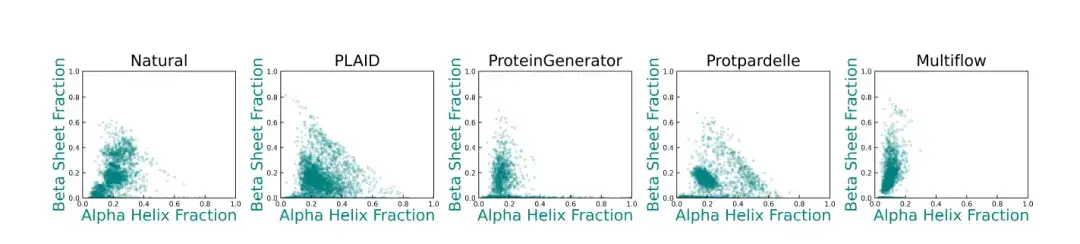
* 이 그림은 천연 단백질의 α-나선과 β-시트 함량 분포와 다양한 방법을 통해 생성된 단백질 구조를 보여줍니다. 각 점은 구조를 나타내며, 좌표는 α-나선형 잔류물의 비율(x축)과 β-시트 잔류물의 비율(y축)을 나타냅니다.
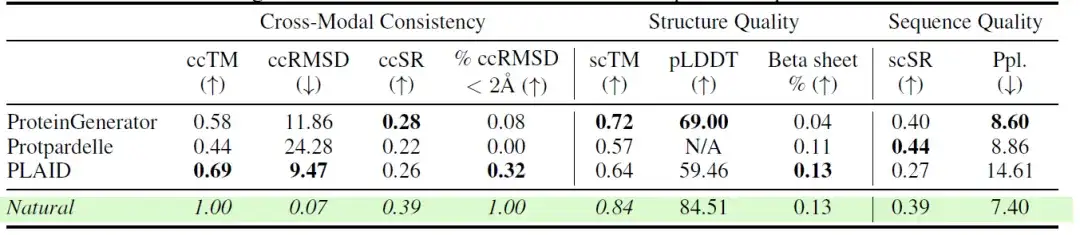
연구자들은 또한 모든 원자 단백질 생성 작업에서 다양한 일관성 및 품질 지표에 걸쳐 다양한 모델의 성능을 비교했습니다. 결과는 다음 표에 나와 있습니다.PLAID로 생성된 샘플은 서열과 구조 간에 높은 교차 모달 일관성을 보여줍니다.
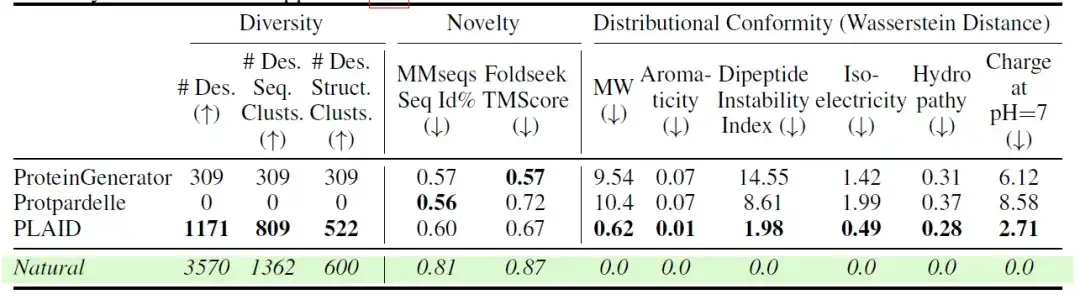
연구자들은 또한 다양한 모델의 다양성, 참신성, 자연스러움을 추가로 평가했습니다. 결과는 다음 표에 나와 있습니다.모든 원자 모델 중에서 PLAID는 시퀀스와 구조 공간 모두에서 가장 독특하고 설계된 샘플을 생성했습니다.
PLAID는 다운스트림의 많은 기능으로 쉽게 확장될 수 있으며 ESMFold에 국한되지 않고 모든 예측 모델에 적용될 수 있다는 점을 강조할 가치가 있습니다.
AI, 단백질 연구의 새로운 길을 열다
확산 변압기는 생물학 분야에서 점점 더 많이 사용되고 있습니다.
이 논문에서는 모델 구축 과정에서 연구자들이 확산 변압기(DiT)를 사용하여 잡음 제거 작업을 수행했다고 언급합니다.
DiT의 기본 원리는 확산 모델에 Transformer 아키텍처를 적용하는 것입니다. 확산 모델은 일반적으로 점진적으로 노이즈를 추가하여 원본 데이터를 손상시킨 다음 모델 학습을 통해 이 데이터를 복구합니다. DiT는 확산 모델에 변환기 블록(적응형 계층 정규화, 교차 주의 등)을 도입하여 모델의 생성 능력을 향상시킵니다.
최근 몇 년 동안 DiT는 이미지 및 비디오 생성 분야에서 상당한 진전을 이루었습니다. Sora와 같은 최첨단 세대 모델의 주요 아키텍처는 DiT입니다.생물의학 분야에서 확산 변압기의 적용은 점점 더 확대되고 있습니다. 이는 연구자들이 잠재적인 약물 분자를 신속하게 선별하고 생물학적 활동을 예측하는 데 도움이 될 수 있습니다. 또한 유전자 서열 분석 및 단백질 구조 예측과 같은 복잡한 작업을 지원하여 생명 과학 연구를 위한 강력한 도구를 제공합니다.단백질 잡음 제거를 예로 들면, DiT는 복잡한 서열-구조 관계를 포착할 수 있습니다. 즉, Transformer의 글로벌 셀프 어텐션 메커니즘을 통해 단백질 서열과 구조 사이의 복잡한 상호 작용을 효과적으로 모델링한 다음 확산 모델의 역과정을 사용하여 각 시간 단계에서 잡음이 제거된 잠재 벡터를 예측하고 잡음에서 단백질의 구조와 서열을 점진적으로 복원할 수 있습니다.
특히 이 논문의 경우 DiT는 혼합된 입력 방식을 처리하기 위한 미세 조정을 위한 보다 유연한 옵션을 제공하며, 특히 단백질 구조 예측 모델이 핵산과 소분자 리간드 복합체를 통합하기 시작함에 따라 이러한 옵션이 더욱 중요해졌습니다. 더욱이 이 접근 방식은 Transformer 교육 인프라를 더 잘 활용할 수 있습니다.
초기 실험에서 연구자들은 삼각형 자기 주의를 사용하는 것보다 사용 가능한 메모리를 더 큰 DiT 모델에 할당하는 것이 더 효율적이라는 것을 발견했습니다. xFormers가 구현한 최적화 알고리즘 학습 모델을 사용하여 추론 단계의 벤치마크 테스트에서 55.8%의 속도 향상과 15.6%의 GPU 메모리 사용량 감소를 달성했습니다.
머신러닝으로 맞춤형 단백질을 '꿈이 현실로'
위에서 언급한 UC 버클리의 연구는 단백질 맞춤화에 있어서 또 다른 중요한 진전이라고 할 수 있습니다. 우리는 단백질이 일반적으로 생명의 구성 요소로 간주될 수 있는 20가지의 아미노산으로 이루어져 있다는 것을 알고 있습니다.수십 년 전만 해도 단백질의 매우 복잡한 구조 때문에 과학자들이 단백질의 3차원 구조를 예측하고 인간이 사용할 수 있는 새로운 단백질을 설계하는 것은 여전히 "꿈"에 불과했습니다. 그러나 최근 몇 년 동안 머신 러닝이 급속히 발전하면서 맞춤형 단백질을 설계하는 꿈이 점차 가능해졌습니다.
잘 알려진 AlphaFold 외에도 주목할 만한 연구 진행 상황은 다음과 같습니다.
2024년 11월, 미국 에너지부 산하 Argonne National Laboratory의 한 팀이 MProt-DPO라는 혁신적인 컴퓨팅 프레임워크를 성공적으로 개발했습니다.이 프레임워크는 인공지능 기술과 세계 최고의 슈퍼컴퓨터를 결합하여 단백질 설계의 새로운 시대를 열었습니다. MProt-DPO를 예로 들면, 과학자들은 특정 조건에서 화학 반응을 효율적으로 촉진할 수 있는 새로운 유형의 효소를 설계했습니다. 기존의 설계 방법과 비교했을 때, 새로운 효소 반응 효율은 약 30%만큼 향상되어 실험 진행 속도가 빨라질 뿐만 아니라 산업적 응용 분야에서 더 많은 가능성을 제공합니다. 또한, MProt-DPO의 성공적인 적용은 항바이러스 단백질의 설계에 대한 새로운 아이디어를 제공합니다. 관련 연구 결과는 "MProt-DPO: 직접 선호도 최적화를 통한 다중 모드 단백질 설계 워크플로우의 ExaFLOPS 장벽 깨기"라는 제목으로 IEEE 컴퓨터 학회에 게재되었습니다.
서류 주소:
https://www.computer.org/csdl/proceedings-article/sc/2024/529100a074/21HUV88n1F6
단백질 포켓은 특정 분자와 결합하기에 적합한 단백질의 부위입니다. 단백질 포켓 디자인은 단백질을 맞춤화하는 과정에서 중요한 방법 중 하나입니다. 2024년 12월, 중국과학기술대학과 협력자들은 딥러닝 알고리즘인 PocketGen을 설계했습니다.단백질 포켓 서열과 구조는 단백질 프레임워크와 결합된 소분자를 기반으로 생성될 수 있습니다. 실험 결과 PocketGen 모델 친화성 및 구조적 합리성과 같은 지표가 기존 방식보다 뛰어나고, 계산 효율성도 크게 향상되었습니다. 관련 연구 결과는 "PocketGen을 이용한 단백질 포켓의 효율적인 생성"이라는 제목으로 Nature Machine Intelligence에 게재되었습니다.
서류 주소:
https://www.nature.com/articles/s42256-024-00920-9

앞으로 인공지능이 단백질 분야에 더욱 응용되면 사람들은 단백질의 공간 구조에 얽힌 비밀을 더욱 깊이 이해할 수 있을 것으로 믿습니다.
참고문헌:
1.https://www.biorxiv.org/content/10.1101/2024.12.02.626353v1
2.https://mp.weixin.qq.com/s/_5_L7bvl-vHtls8gBbfSmQ
3.https://mp.weixin.qq.com/s/sfrm2rj_8kH0JA2vu4NmTw
4.http://www.news.cn/globe/20241014/f7137840e56340f081f9eb819d87ba40/c.html
5.http://www.bfse.cas.cn/yjjz/202412/t20241212_5042432.html
6.https://www.sohu.com/a/826241274_12