Command Palette
Search for a command to run...
상하이 교통대학교의 Zhong Bozitao가 AlphaFold 3의 완전한 분해를 설명했습니다. 원자 정밀도로 모든 생체 분자 구조를 예측하기 위해 데이터를 극단적으로 사용했지만 완벽하지는 않습니다.

모든 생체 분자의 구조와 상호작용을 "원자 정밀도"로 예측할 수 있는 AlphaFold 3는 출시되자마자 업계에서 폭넓은 논의를 불러일으켰습니다. 8월 13일, 상하이 교통대학교 AI 생명공학 여름학교에서중보즈타오 박사는 "알파폴드 3: 원리, 응용 및 전망"이라는 주제로 자신의 학습 경험을 체계적으로 정리했으며, 과학 연구 커뮤니티에서 많은 관련 연구 결과를 광범위하게 정리하여 알파폴드 3에 대한 그의 심오한 통찰력을 모든 사람과 공유했습니다.HyperAI는 원래 의도를 훼손하지 않으면서 연설의 핵심 내용을 구성했습니다. 다음은 연설 전문입니다.

오늘은 단백질 구조 예측에 초점을 맞춰, 현재 최고의 단백질 구조 예측 도구이자 더욱 광범위한 생물학적 분자 구조 예측 도구인 AlphaFold 3에 대해 알아보겠습니다.AlphaFold 3의 상태는 자명합니다.
단백질 합성은 DNA 전사로 시작하며, 전사는 유전 정보를 RNA로 전달하고, RNA는 다시 단백질로 번역되며, 나아가 2차, 3차, 4차 구조로 접힙니다. 대부분의 단백질은 고유한 형태로 접히며, 구조에 필요한 정보는 아미노산 서열에 인코딩되어 있습니다. 이를 우리는 종종 "서열은 구조를 결정하고, 구조는 기능을 결정한다"고 말합니다.단백질 구조 예측은 생물학적 기능을 이해하는 데 매우 중요합니다.
AlphaFold 3 혁신: 혁신적인 모델 아키텍처와 향상된 데이터 활용
AlphaFold 3와 AlphaFold 2 모델 아키텍처 비교
과거에는 AlphaFold 2가 단백질 구조 예측에서 다른 알고리즘을 직접 "이겼습니다".핵심 아키텍처는 아래 그림과 같이 세 가지 주요 부분으로 요약할 수 있습니다. 첫 번째 부분인 파란색 상자의 MSA 및 템플릿 모듈은 모델의 입력 데이터로 다중 시퀀스 정렬(MSA) 및 템플릿 구조 정보를 수집하고 통합하는 역할을 합니다. 두 번째 부분인 녹색 상자의 Evoformer 모듈은 다중 시퀀스 조직의 공진화 정보를 이해하고, 수집된 정보를 정제하고 처리하여 세 번째 부분의 보라색 상자에 있는 구조 모듈로 전달하는 역할을 합니다.
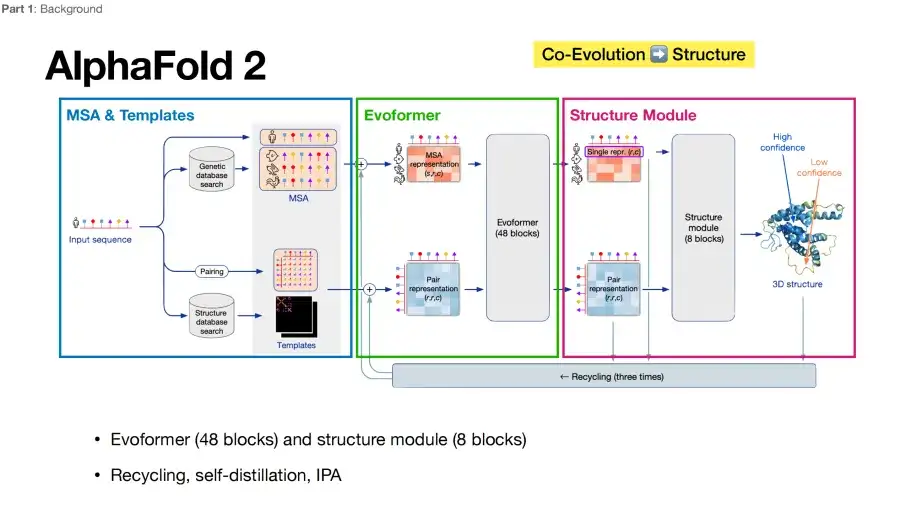
딥러닝 관점에서 볼 때, Evoformer는 인코더 역할을 하고, Structure Module은 디코더와 같은 역할을 합니다.AlphaFold 2는 시퀀스 입력에서 구조적 출력으로 직접 매핑하는 엔드투엔드 최적화 기능으로 인해 많은 호평을 받았습니다.
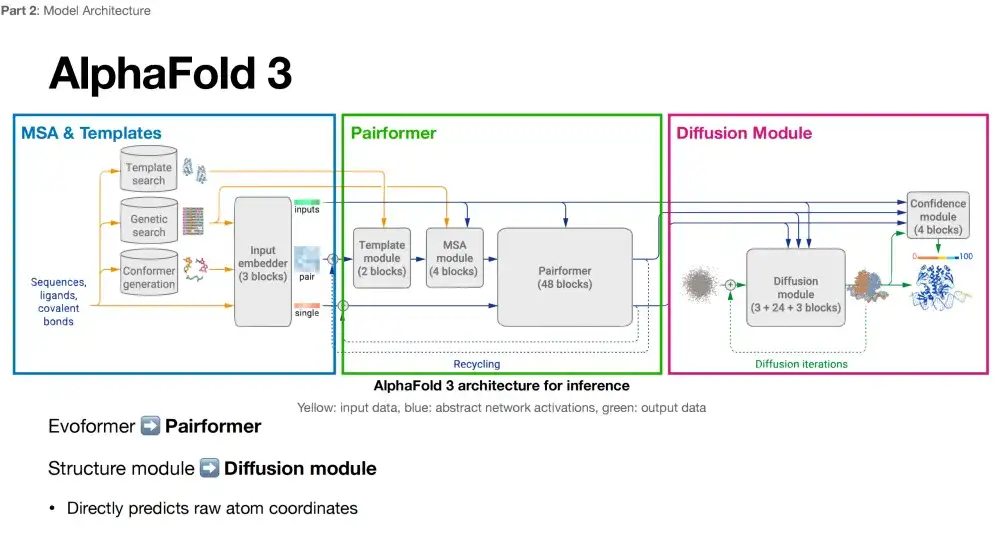
AlphaFold 3의 모델 아키텍처의 변화는 상상만큼 크지 않을 것이라는 의견이 일반적이다. 이 모델 프레임워크는 3가지 핵심 부분으로 구성되어 있습니다. 각 부분과 AlphaFold 2를 비교한 내용은 다음과 같습니다.
1부: 매우 유사하게 유지하기
아래 그림에서 볼 수 있듯이 AlphaFold 3와 AlphaFold 2의 아키텍처 다이어그램을 비교하면 AlphaFold 3의 첫 번째 부분(파란색 상자)에는 여전히 MSA 및 템플릿이 포함되어 있으며, 추가로 Conformer 생성 링크가 도입된 것을 알 수 있습니다.
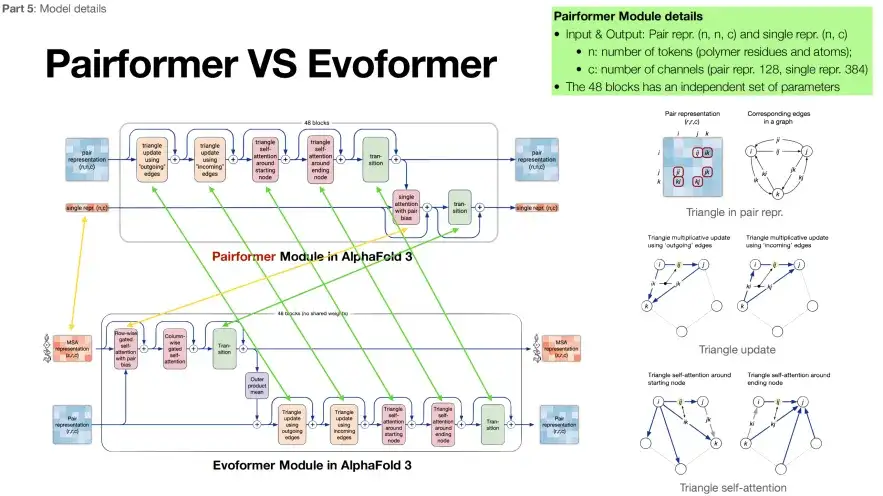
2부: MSA 시퀀스에 대한 의존도 감소
AlphaFold 3의 두 번째 부분(녹색 상자 안)은 Pairformer라고 합니다. 구조는 기본적으로 Evoformer와 매우 유사하지만 MSA 모듈의 수는 4개로 줄었습니다. 아래 그림에서 볼 수 있듯이 녹색 화살표는 두 모듈의 동일한 내용을 나타내고 노란색 화살표는 차이점을 나타냅니다. 보시다시피,AlphaFold 3는 표적 단백질 서열에 더 많은 중점을 두고 MSA 서열에는 덜 의존합니다.
또한, AlphaFold 3가 여러 작업에서 강력한 성능을 보일 수 있는 이유는 다중 시퀀스 정렬(MSA)에 대한 의존도를 낮추기 때문일 수 있다고 생각합니다.아래 그림에서 볼 수 있듯이 오른쪽은 MSA가 AlphaFold 2 성능에 미치는 영향을 보여줍니다. MSA의 수가 늘어나고 특정 임계값(분홍색 선)을 넘으면 AlphaFold 2 성능의 향상이 안정화되는 경향을 보입니다. 아래 그림의 중간 부분에서 볼 수 있듯이, AlphaFold 2에 비해 AlphaFold 3에 대한 MSA의 영향은 약해졌습니다(곡선의 변동이 매우 적습니다).
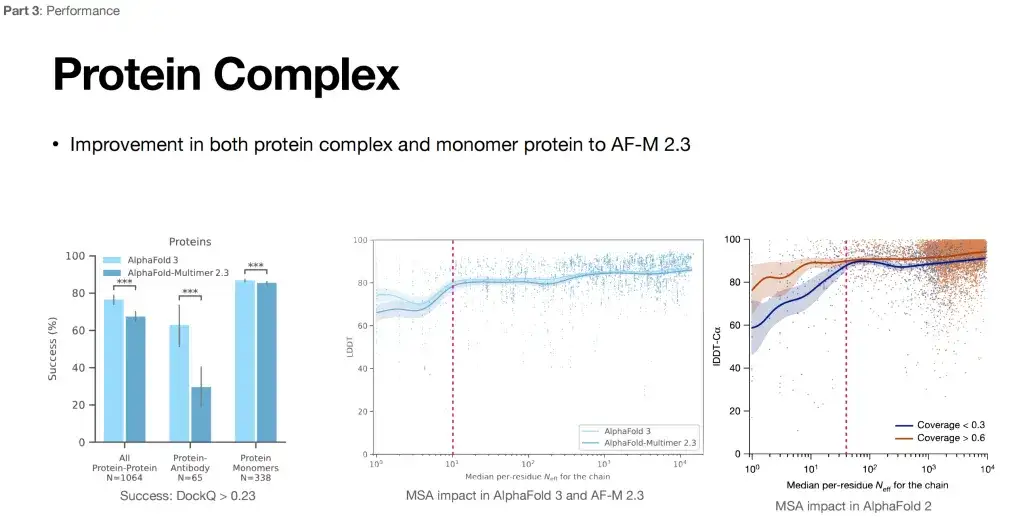
또한 항체의 성숙에는 종종 생체 내 과돌연변이 과정이 필요합니다. MSA 정보는 단백질의 구조를 예측하는 데 큰 도움이 되지 않으며, 단백질과 그 복합체에 대한 MSA 정보를 짝지어 찾는 것도 어렵습니다. 이러한 관점에서 볼 때, AlphaFold 3의 적용 범위 확대는 MSA에 대한 의존도를 낮추는 것으로 볼 수 있다.
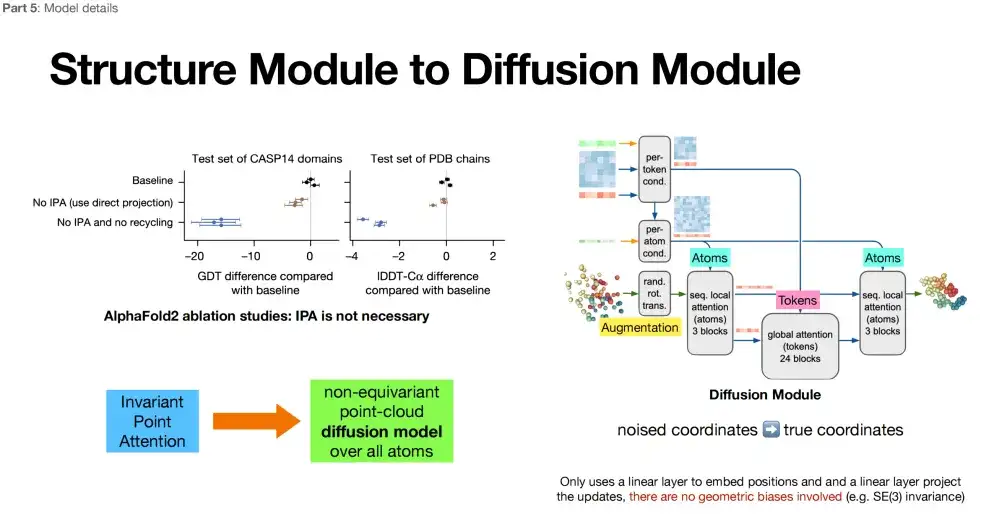
3부: 모든 원자 구조 생성 + 스테레오 회전 불변성 제거
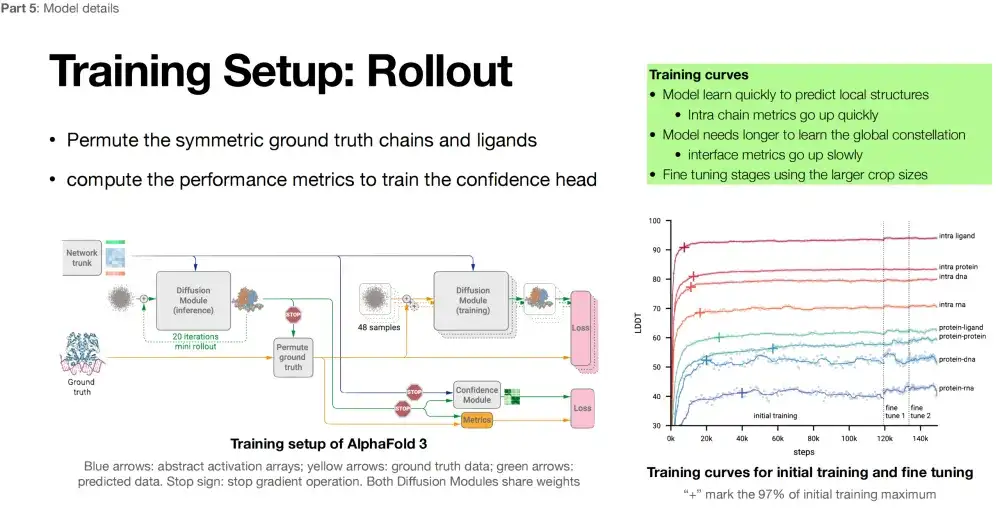
AlphaFold 3의 세 번째 부분(보라색 상자)은 구조 모듈 범주에 속하는 확산 모델을 채택했습니다. 차이점은 확산 모델은 구조 모듈의 반복적인 최적화를 확산 모델이라는 새로운 메커니즘으로 대체한다는 것입니다.
*확산 모델: 모델에 노이즈를 추가(전방), 모델에서 노이즈를 제거(역방)한 후 역방향 프로세스를 학습하여 유사한 데이터 분포를 생성합니다.
아래 그림에서 볼 수 있듯이, 세 번째 부분에서 AlphaFold 3는 모든 원자 수준에서 구조 생성을 달성합니다. 분자의 기본 구성 요소인 원자는 더 풍부한 물리적 정보를 담고 있을 수 있으며, 이는 AlphaFold 3가 단백질 구조를 예측할 때 더 심층적인 물리 법칙을 포착할 수 있음을 의미합니다. 또한 AlphaFold 3에서는 AlphaFold 2에서 강조되었던 스테레오 회전 불변성을 버렸습니다. AlphaFold 2에서 이 기능의 추가 아키텍처를 삭제한 후, 연구진은 모델(확산 모듈)의 설계가 더 자유로워졌다는 것을 발견했습니다.
AlphaFold 3는 데이터 활용도를 향상시킵니다.
단백질 데이터 리소스는 제한적이지만, AlphaFold 3는 데이터 세트를 더 크게 만들 뿐만 아니라 데이터 활용도도 향상시킵니다.구체적으로 AlphaFold 2의 백만 수준 데이터 세트와 비교했을 때, AlphaFold 3는 십억 수준에 직접 접근하며, 학습 세트도 더 큽니다. 또한 PDB의 데이터 외에도 해당 학습 세트에는 많은 양의 다른 데이터가 통합되어 있습니다. 예를 들어, AlphaFold 2가 예측한 구조적 데이터는 훈련 세트를 확장하면 더욱 정확해집니다. 구체적인 훈련 세트는 아래 그림과 같습니다.

AlphaFold 3는 적용 범위에서 엄청난 도약을 이뤘습니다.
AlphaFold 3의 가장 큰 변화는 적용 범위에서 질적인 도약을 이루었다는 점입니다.과거에는 AlphaFold 2가 아미노산 구조를 예측하는 데 더 중점을 두었지만, AlphaFold 3는 원자 수준의 구조를 직접 예측할 수 있습니다. 기능적 확장은 구체적으로 다음 네 가지 측면에서 반영됩니다.
* 리간드를 정확하게 예측할 수 있습니다. 즉, 단백질 내 소분자의 결합 부위를 예측할 수 있습니다.
* 단백질 복합체 구조를 예측할 수 있음
* 단백질과 핵산의 번역 후 변형 구조를 예측할 수 있습니다.
* DNA와 RNA의 구조, DNA/RNA 복합체와 단백질의 구조를 예측할 수 있습니다.
AlphaFold 3는 리간드 도킹 분야를 변화시킵니다.
그 중에서도 AlphaFold 3가 과학 분야에 미친 가장 큰 영향은 리간드 도킹 작업의 향상입니다.아래 그림에서 볼 수 있듯이, PostBusters Benchmark를 통해 4가지 리간드 도킹 작업에서 다양한 딥러닝 알고리즘의 성공률을 평가했습니다. 알려지지 않은 포켓과 구조적 사전 지식이라는 전제 하에 AlphaFold 3가 76.4%라는 가장 높은 성공률을 달성할 수 있음을 알 수 있습니다.
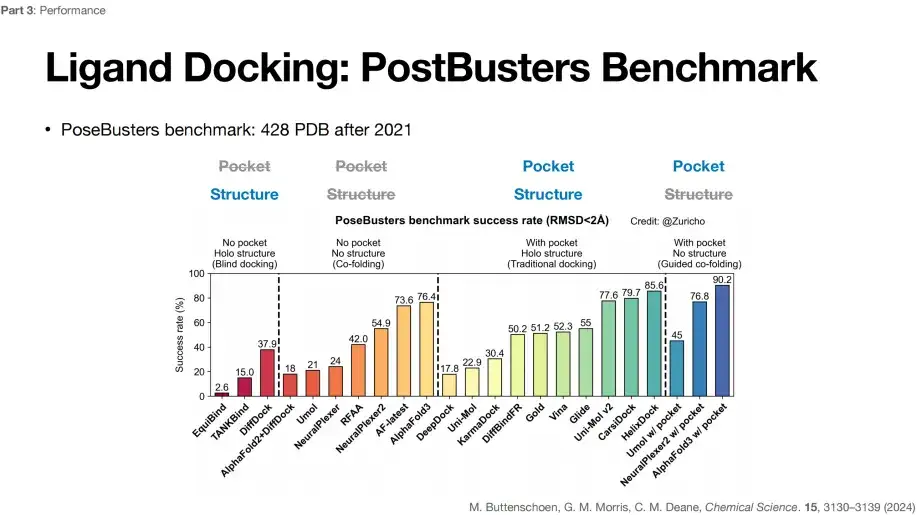
PostBusters Benchmark는 2021년 이후 428개 PDB 데이터를 선택합니다.
작업 성공의 기준은 예측된 소분자 도킹 위치와 실제 도킹 위치의 편차가 2Å 미만이라는 것입니다.
위 그림에서 보듯이, 포켓 위치가 알려지지 않고 단백질 구조가 알려진(포켓 없음, 홀로 구조) 첫 번째 유형의 블라인드 도킹 작업에서 DiffDock은 37.9%의 가장 높은 성공률을 달성할 수 있습니다.
두 번째 유형의 공동 접힘 작업(소형 분자 및 단백질 구조가 접힘)에서는 포켓 위치가 알려지지 않고 단백질 구조도 알려지지 않은 경우(포켓 없음, 구조 없음) AlphaFold 2+DiffDock 결합 예측의 성공률이 18%로 떨어졌습니다.또한 AlphaFold 3는 가장 높은 76.4% 성공률을 달성했는데, 이는 AlphaFold 3가 정확하게 예측할 뿐만 아니라 주머니와 구조에 대한 사전 지식에 의존하지 않는다는 것을 보여줍니다.
세 번째 범주의 전통적인 도킹 작업에서는 소분자의 포켓 위치와 단백질 구조가 알려져 있습니다(포켓 포함, 홀로 구조). 즉, 포켓이 노출되어 있습니다. Gold는 51.2%의 성공률을 달성했고, Vina는 52.3%의 성공률을 보였으며, Glide는 55%로 증가했습니다. 다른 딥러닝 알고리즘도 비교적 좋은 수준에 도달할 수 있는데, 이는 성공률이 포켓의 영향을 받는다는 것을 나타냅니다.
포켓 위치가 알려져 있고 단백질 구조가 알려지지 않은(포켓 있음, 구조 없음) 가이드 공동 접힘 작업의 네 번째 범주에서 모델의 성공률은 AlphaFold 3의 경우 76.4%에서 90.2%로 크게 향상되었습니다. 이는 알려진 포켓 정보가 작업의 성공률을 높일 수 있음을 나타냅니다. 그러나 포켓의 정의에 대해서는 논란이 있습니다.따라서 리간드 도킹 과제에 대한 AlphaFold 3의 구체적인 개선 사항을 알고 싶다면, 상대적으로 더 안정적인 두 번째 유형의 과제의 성공률만 고려하면 됩니다.
아래 그림에서 볼 수 있듯이, 다양한 모델 간에 포켓 정의에 상당한 차이가 있습니다. Gold의 포켓은 25Å 구(그림의 왼쪽 상단 모서리에 있는 파란색 부분)인 반면, Vina 모델은 포켓 표현으로 25Å 큐브를 사용하고, DeepDock 포켓 크기는 10Å, Uni-Mol 포켓 크기는 8Å입니다.
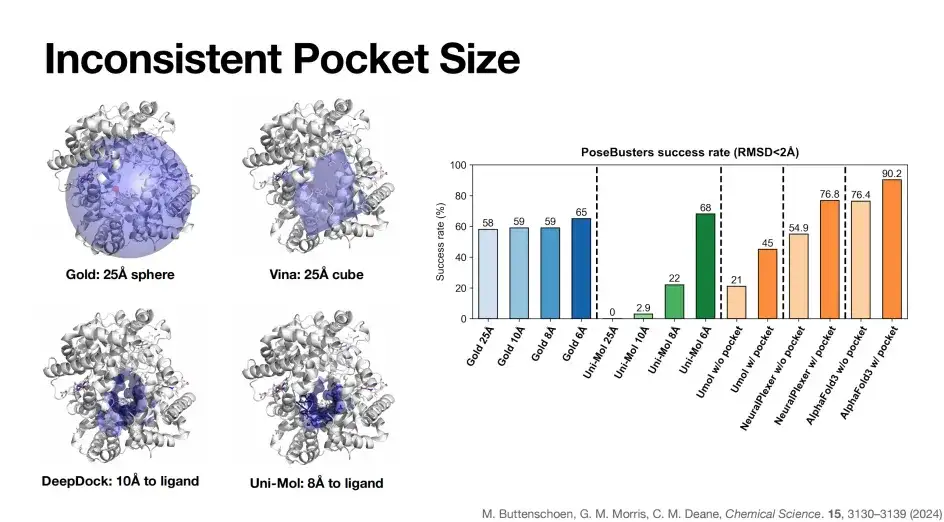
위 그림의 오른쪽에서 볼 수 있듯이 Gold 모델의 포켓 크기가 25Å에서 6Å로 점차 줄어들었을 때 PoseBusters 벤치마크 성공률은 비교적 안정적으로 나타났습니다. 이는 Gold의 물리 기반 알고리즘의 특성 때문입니다. 이와 대조적으로, 딥러닝 알고리즘인 유니몰은 포켓을 점차 6Å로 좁혔고, 성공률은 68%로 증가한 후 25Å에서 0으로 떨어졌는데, 이는 일부 딥러닝 도킹 알고리즘이 포켓에 의존한다는 것을 반영합니다.
마찬가지로 앞서 언급했듯이 포켓 정보가 도입된 이후 AlphaFold 3의 도킹 성공률은 76.4%에서 90.2%로 크게 향상되었습니다.요약하자면, 포켓 정보는 모델 예측의 성공률을 높이는 데 중요한 역할을 합니다. 하지만 이상적으로는 포켓이나 구조적 정보 없이도 높은 정확도를 달성할 수 있는 모델, 즉 AlphaFold 3가 최선의 선택이 될 것입니다.
AlphaFold 3는 항체 및 항원 구조 예측을 가능하게 합니다.
AlphaFold 3의 또 다른 응용 분야는 항체와 항원의 구조 예측입니다. 아래 그림의 왼쪽은 항체 및 항원 구조 예측을 위한 AlphaFold 3의 성능 평가를 보여줍니다. 더 낮은 평가 기준(DockQ>0.23)에서는 단 한 번의 시도만으로 AlphaFold 3의 예측 성공률은 40%(하늘색 선)보다 낮지만, 1,000번 시도 후에는 예측 성공률을 60%까지 향상시킬 수 있습니다.
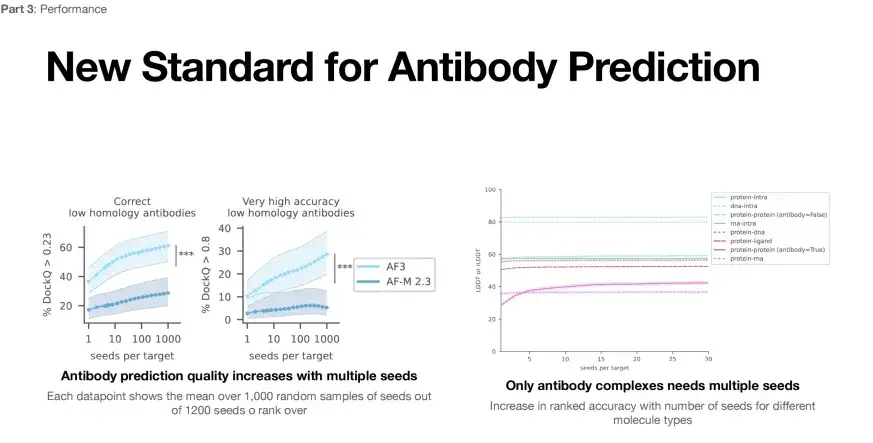
* 왼쪽: 항체 구조 예측, 각 데이터 포인트는 1,200개의 씨앗 중 무작위로 선택된 1,000개의 씨앗의 평균 점수를 나타냅니다.
* 오른쪽: 평가지수 DockQ가 0.23보다 큰 경우 구조적 정확도가 아직 검증되지 않은 것으로 간주할 수 있습니다. DockQ가 0.8을 초과하면 구조 예측이 매우 정확합니다.
더욱이, 더 엄격한 기준(DockQ>0.8)으로 측정할 경우, 단일 실행의 성공률은 10% 정도로 낮을 수 있으며, 실행 횟수를 1,000회로 늘리면 성공률은 30%까지 높아질 수 있습니다.이는 AlphaFold 3 실행 횟수(대상당 시드)를 늘리면 항체 항원 구조 예측의 성공률을 높일 수 있음을 보여줍니다.
하지만 위 그림의 오른쪽에서 볼 수 있듯이 AlphaFold 3는 단백질-단백질 복합체의 구조를 예측할 때 실행 횟수를 늘려야만 성공률을 높일 수 있습니다. 이는 다른 유형의 복잡한 구조를 예측하는 데 AlphaFold 3를 적용하는 데도 추가적으로 최적화가 필요하다는 것을 보여줍니다.
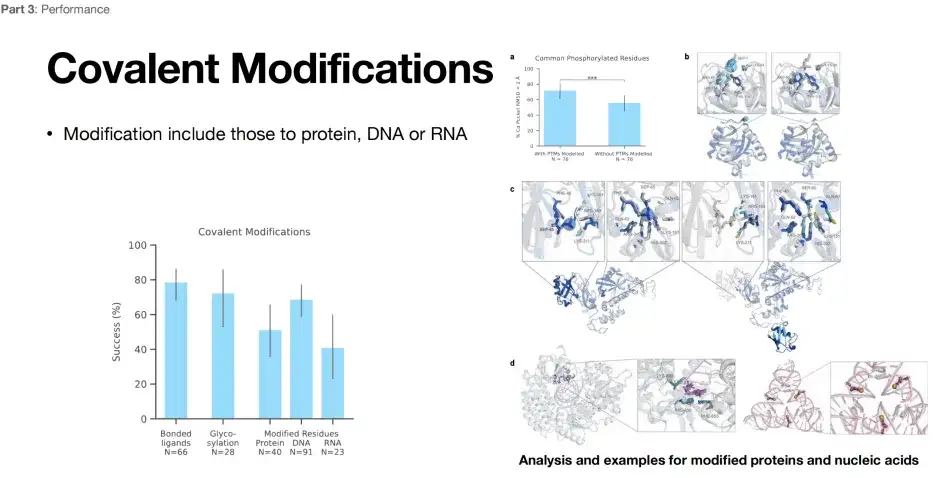
AlphaFold 3는 공유 결합 변형 예측을 가능하게 합니다.
아래 그림에서 볼 수 있듯이 AlphaFold 3는 변형 예측 측면에서도 뛰어난 구조적 예측 능력을 보여줍니다.성공률은 약 80%, 60%, 40%에 도달할 수 있습니다. AlphaFold 3는 공유 결합 변형에 참여하는 연구자들에게 의심할 여지 없이 강력한 도구입니다.
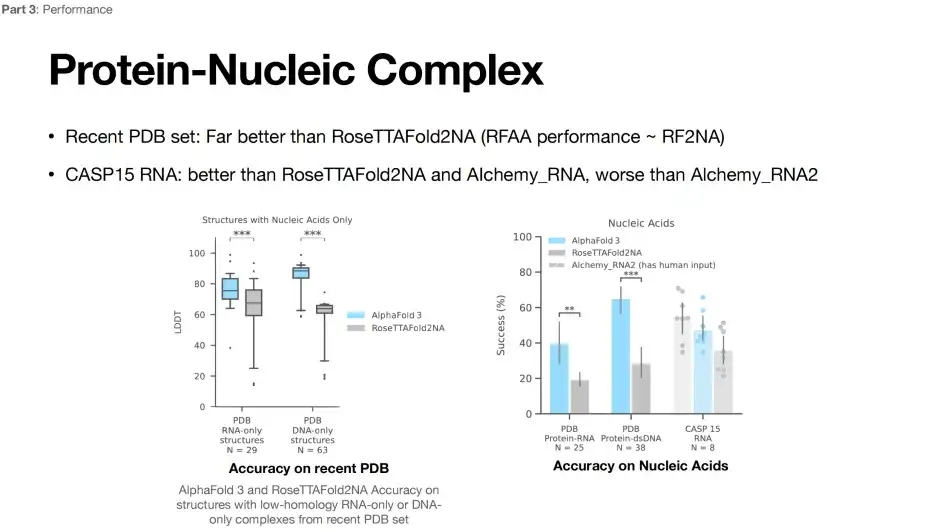
RNA 구조 예측에 있어 AlphaFold 3의 한계
현재로선 RNA 구조를 예측하는 것은 여전히 어려운 일이다.아래 그림에서 볼 수 있듯이 AlphaFold 3는 RoseTTAFoId2NA 모델에 비해 예측 성능이 크게 향상되었습니다. 그러나 CASP15 RNA 구조를 예측할 때 AlphaFold 3의 정확도는 Alchemy_RNA2(인간의 입력이 있음) 모델보다 낮습니다.
다양한 작업에 대한 AlphaFold 3의 장단점 비교
AlphaFold 3의 학습 곡선을 분석하면 다양한 작업에서 모델의 성능을 명확하게 확인할 수 있습니다. LDDT 지표가 높을수록 좋습니다.아래 그림에서 볼 수 있듯이, 이 모델은 리간드 내부 구조를 예측하는 데 가장 좋은 성능을 보입니다. 또한 단백질 내부 구조를 예측하는 데 높은 정확도를 보여줍니다. 이 모델은 DNA의 안정적인 이중나선 구조 덕분에 DNA 내부 구조를 예측하는 데도 좋은 성능을 보입니다. 이와 대조적으로, 이 모델은 RNA 내부 구조를 예측하는 데는 성능이 좋지 않습니다.
복합체 예측 분야로 눈을 돌리면, 이 모델은 단백질-리간드 복합체 구조 예측에서 가장 좋은 성과를 보였고, 그 다음으로 단백질-단백질 복합체 예측에서 좋은 성과를 보였습니다. 단백질-DNA 복합체 예측에서는 모델 성능이 떨어졌고, 단백질-RNA 복합체 예측에서는 가장 나쁜 성능을 보였습니다. 이 결과는 RNA 구조 예측의 어려움을 반영합니다. RNA 구조 데이터는 부족하고, 그 구조는 역동적이고 유연합니다. 이는 구조 생물학 분야가 현재 직면한 과제 중 하나입니다.
또한 연구자들은 구조 예측을 위해 AlphaFold 3를 사용했을 때,예측 결과의 신뢰성은 PAE 표를 통해서도 평가할 수 있습니다.

AlphaFold 3는 완벽하지 않습니다
AlphaFold 3는 완벽하지 않습니다. 예를 들어, 잘못된 키랄성을 선택할 수도 있습니다.작업 중에 비정상적인 상황이 발생하면 결과의 안정성을 확인하기 위해 테스트를 여러 번 실행하는 것이 좋습니다. 두 번째로, AlphaFold 3 역시 단백질 동역학을 예측하는 데 한계가 있는데, 이는 구조적 데이터가 부족하고 단백질의 다차원적 구조 정보를 파악할 수 없기 때문일 수 있습니다.
*어떤 물체가 거울상과 다를 경우, 그 물체는 "카이랄"이라고 하며, 그 거울상은 원래 물체에 겹쳐질 수 없습니다. 마치 왼손과 오른손이 서로의 거울상이어서 겹쳐질 수 없는 것과 같습니다.
또한 AlphaFold 3는 생성 모델에서 흔히 발생하는 문제인 환각이라는 문제점도 가지고 있습니다.아래의 단백질 구조 예측 결과에서 볼 수 있듯이, 왼쪽 단백질 구조의 회색 부분만 해소될 수 있고, 나머지는 전자 밀도가 부족하여 펼쳐진 상태일 수 있습니다. 가운데 그림은 AlphaFold 2가 단백질을 예측한 결과입니다. 파란색 부분은 접힌 상태로 간주되고, 나머지 "리본" 부분은 펼쳐진 상태로 간주됩니다. 예측된 구조는 비교적 합리적이다. 오른쪽은 모든 접을 수 있는 영역을 접는 경향이 있는 AlphaFold 3의 예측 결과입니다. 이런 구조는 타당해 보이지만 실제로 위의 영역 대부분은 실제로 접혀 있지 않습니다.따라서 AlphaFold 3의 환상은 단백질이 펼쳐진 상태를 그대로 유지하는 대신 접힌 상태로 예측하는 경향이 있습니다.

AlphaFold 3의 환각 문제를 해결하기 위해연구진은 직접적이고 효과적인 방법을 선택했습니다. AlphaFold 2에서 예측한 결과가 비교적 합리적이었기 때문에 AlphaFold 2에서 예측한 결과를 AlphaFold 3의 학습 데이터 세트에 통합하여 모델의 학습 효과를 향상시켰습니다. 하지만 이 방법에는 한계가 있습니다. AlphaFold 2 자체의 예측에 오류가 있는 경우, 다른 데이터 소스를 도입하여 모델을 더욱 최적화하지 않는 한 AlphaFold 3의 예측 품질에 영향을 미칠 수 있습니다.
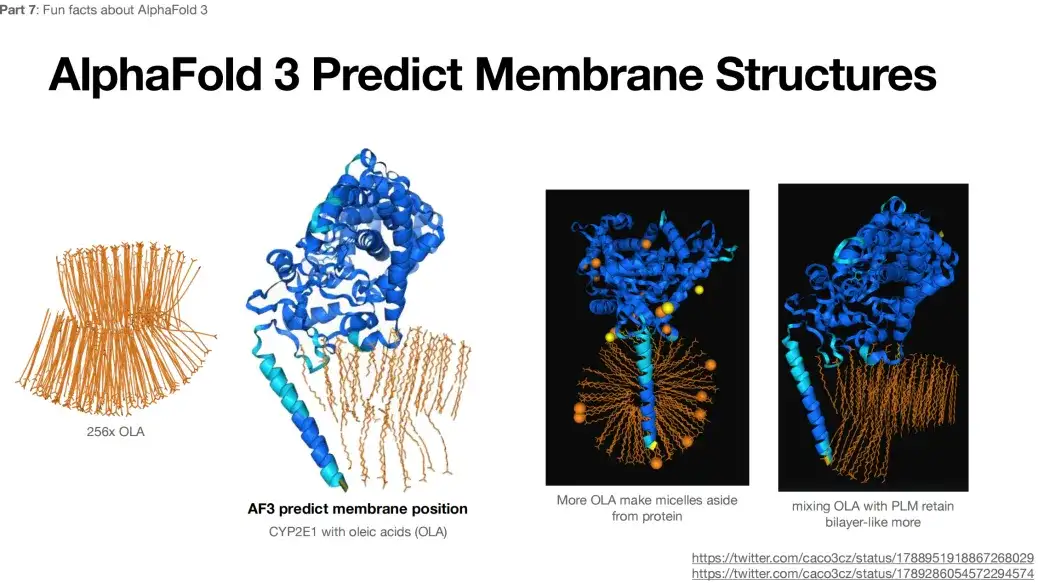
또한, 256x OLA가 AlphaFold 3에 입력으로 제출되면 예측된 결과는 아래 그림과 같이 이중층과 같은 구조를 보여줍니다.이런 구조는 예상되거나 일반적이지 않습니다.
또한 AlphaFold 3는 RNA와 DNA의 구조를 예측하는 데 정확하지 않습니다.아래 그림에서 보듯이, RNA 구조를 예측할 때 G:G, G:A 등과 같이 이상한 상보적 쌍도 나타납니다.
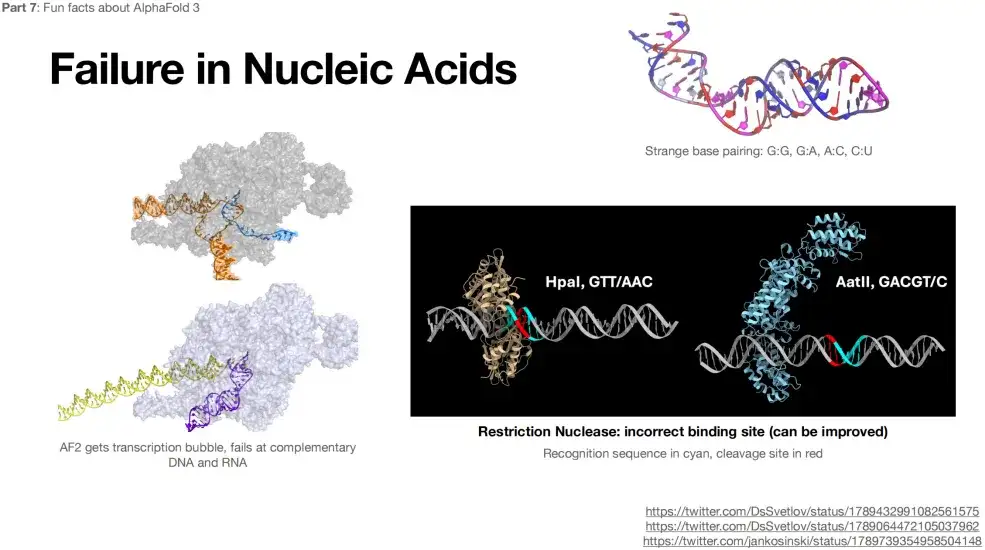
AlphaFold 3 사용의 제한 사항
데이터가 높은 수준의 기밀이 아니라는 전제 하에, 누구나 Google에서 제공하는 웹사이트를 통해 AlphaFold 3에 접근할 수 있습니다. 하지만 이 플랫폼에도 몇 가지 한계가 있습니다. 아래 그림에서 볼 수 있듯이, 단백질 변형 측면에서 AlphaFold 3는 현재 특정 3개 위치에서 제한된 수의 변형만을 지원하여 총 23가지 유형을 지원합니다. DNA 변형은 9가지 유형만 지원하고, RNA 변형은 15가지 유형만 지원하며, 금속 이온은 10가지 금속만 지원하고, 리간드는 14가지 소분자로 제한됩니다.
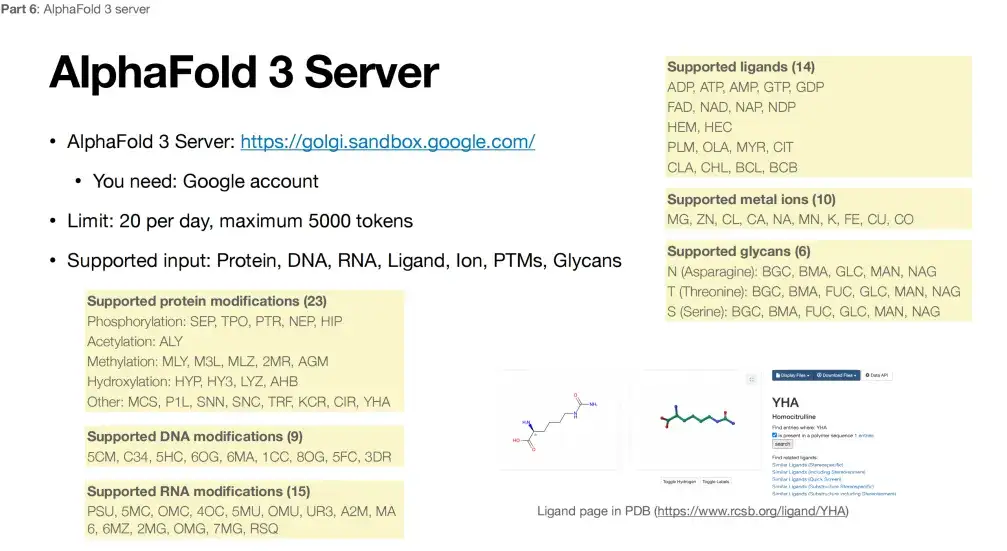
따라서 위에서 언급한 구체적인 한계를 감안할 때 AlphaFold 3는 대부분의 연구와 반응을 처리하지 못할 수 있으며, 진정한 오픈 소스가 될 때까지 기다려야 할 수도 있습니다.
요약하자면, AlphaFold 3는 예측 범위를 확장하여 기존 AI 모델을 능가하는 측면에서 상당한 성과를 거두었지만, 특히 미세 구조 예측 분야에서 특정 작업에 대한 성능은 여전히 개선의 여지가 있습니다.따라서 AlphaFold 3가 상당한 진전을 이루긴 했지만, 특정 복잡한 문제를 완벽하게 해결하기 위해서는 여전히 지속적인 연구와 노력이 필요합니다.
중보쯔타오 소개

종보쯔타오는 현재 상하이 교통대학교에서 인공지능 분야 박사과정을 공부하고 있습니다. 그의 주요 연구 방향으로는 고처리량 단백질 구조 및 기능 예측, 단백질 형태 생성 등이 있습니다. 그는 2019년부터 20편 이상의 논문을 발표했으며, 심해 프로테옴과 대사 경로 간의 관계에 대한 고처리량 AlphaFold 구조 예측 분석 결과를 Nature Communications에 게재했습니다. 그는 국제 유전자 조작 기계 경연대회(iGEM)에서 금메달을 세 번이나 수상했으며, 이 대회의 심사위원으로도 여러 번 활동했습니다.
구글 학술 검색:
https://scholar.google.com/cita