Command Palette
Search for a command to run...
ICML에 선정되었습니다! 인민대학교 연구팀은 등가 그래프 신경망을 사용하여 표적 단백질 결합 부위를 예측했으며, 20%의 성능이 가장 크게 향상되었습니다.
생명체 내에서 거의 모든 생물학적, 약리학적 과정에는 수용체(표적 단백질)와 리간드(소분자) 간의 상호작용이 포함됩니다. 이러한 상호작용은 표적 단백질 구조의 특정 영역에서 발생합니다."결합 부위"로 알려진 이 분야는 표적 단백질의 결합 부위를 예측하는 분야로, 약물 발견과 같은 다양한 하위 작업에서 기본적인 역할을 합니다.
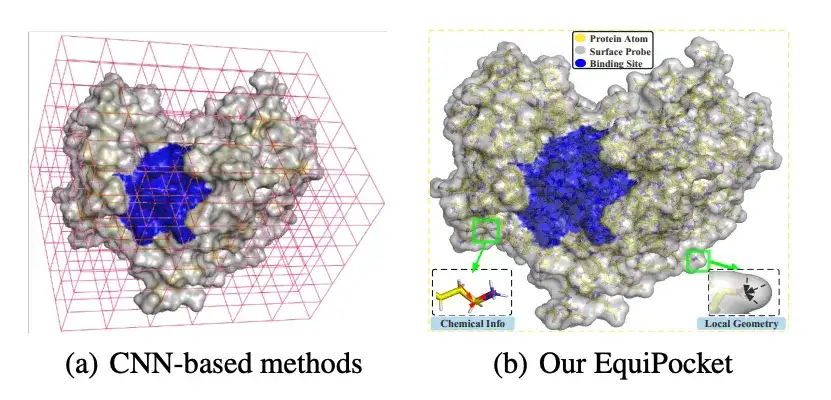
최근 몇 년 동안 딥 러닝의 획기적인 발전에 영감을 받아 합성 신경망(CNN)이 리간드 결합 부위 예측에 성공적으로 적용되었습니다. CNN 기반 방법은 단백질의 원자를 가장 가까운 폭셀로 공간적으로 클러스터링하여 단백질을 3D 이미지로 처리한 다음, 결합 부위 예측을 3D 그리드의 객체 감지 문제 또는 의미 분할 작업으로 모델링합니다. 이러한 방법에는 몇 가지 장점이 있지만 여전히 다음과 같은 과제가 있습니다.불규칙한 단백질 구조를 표현하는 데 결함이 있습니다. 회전에 민감합니다. 이는 단백질 표면의 기하학적 특징을 적절하게 설명하지 못했습니다. 그리고 단백질 크기의 변화에 민감하지 않습니다.
이를 위해 중국 런민대학교 가오링 인공지능학원 연구팀은 최근 AI 분야 최고 학술대회인 ICML 2024에서 "EquiPocket: 리간드 결합 부위 예측을 위한 E(3)-등가 기하 그래프 신경망"이라는 제목의 연구 논문을 발표했습니다. 본 연구는 E(3) 등변 그래프 신경망(GNN)을 리간드 결합 부위 예측에 적용한 최초의 연구이다.EquiPocket이라는 프레임워크를 제안했습니다.CNN 기반 방법에서 발생하는 문제점을 해결합니다.
연구 하이라이트:
* 리간드 결합 부위 예측을 위한 E(3) 등변형 GNN의 첫 번째 응용
* 기존 CNN 기반 방식과 비교했을 때, EquiPocket은 폭셀화가 필요 없고, 불규칙한 단백질 구조를 모델링할 수 있으며, 유클리드 변환에 민감하지 않아 "불규칙한 단백질 구조 표현의 결함" 및 "회전에 대한 민감성"과 같은 문제를 해결합니다.
* 대표적인 벤치마크 방법에 대한 광범위한 실험은 EquiPocket이 현재 최첨단 방법보다 우수함을 보여주며 이는 약물 발견과 같은 다양한 하류 작업에 도움이 될 수 있습니다.
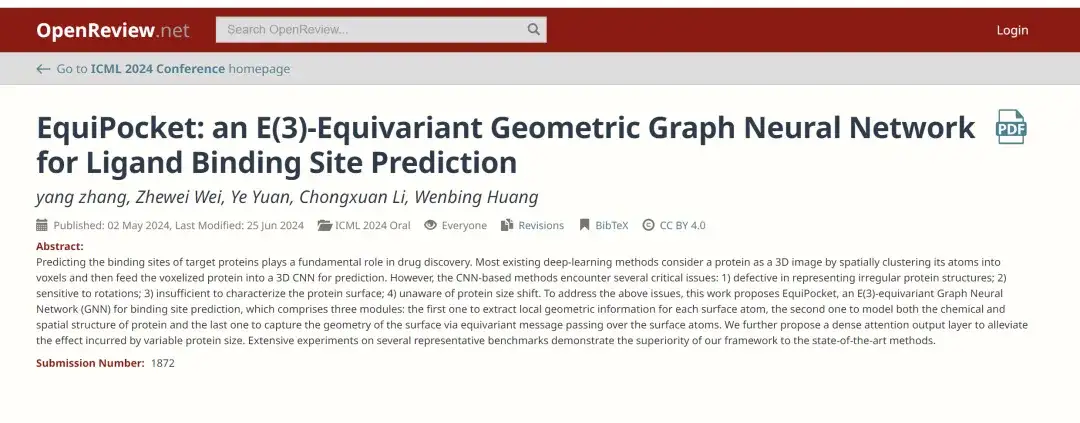
서류 주소:
https://openreview.net/forum?id=1vGN3CSxVs
오픈소스 프로젝트인 "awesome-ai4s"는 100개가 넘는 AI4S 논문 해석을 모아 방대한 데이터 세트와 도구를 제공합니다.
https://github.com/hyperai/awesome-ai4s
데이터 세트: 다양한 전문 데이터 세트의 종합적 검증
연구진은 여러 개의 특수 데이터 세트를 선택하여 결합 부위 예측을 위한 관련 리간드를 포함하는 각 데이터 세트의 mlig 하위 세트를 사용하여 이를 평가했습니다.
안에,scPDB는 결합 부위 예측을 위한 잘 알려진 데이터 세트입니다.VolSite에서 생성된 단백질, 리간드 및 3D 공동 구조가 포함되어 있습니다. 이 연구에서는 17,594개 구조, 16,034개 항목, 4,782개 단백질, 6,326개 리간드를 포함하는 2017년 릴리스를 사용하여 훈련 및 교차 검증을 수행했습니다.
PDBbind는 단백질-리간드 복합체를 연구하는 데 일반적으로 사용되는 데이터 세트입니다.단백질의 3D 구조, 리간드, 결합 부위와 정확한 실험실에서 결정된 결합 친화도 결과가 포함되어 있습니다. 본 연구에서는 평가를 위해 2020년 버전을 사용했는데, 이는 두 부분, 즉 공통 세트(14,127개 복합체)와 개선된 세트(5,316개 복합체)로 구성되어 있습니다. 일반 세트에는 모든 단백질-리간드 복합체가 포함되어 있으며, 개선된 세트는 일반 세트에서 실험 테스트를 위해 더 나은 품질의 화합물을 선택합니다.
COACH 420과 HOLO4K는 결합 부위 예측에 사용되는 두 가지 테스트 데이터 세트입니다.(Krivák & Hoksza, 2018)이 처음 소개했습니다.
모델 아키텍처: EquiPocket의 전체 프레임워크는 세 가지 모듈로 구성됩니다.
EquiPocket의 전체 프레임워크는 3개의 모듈로 구성됩니다.다음 그림과 같이:
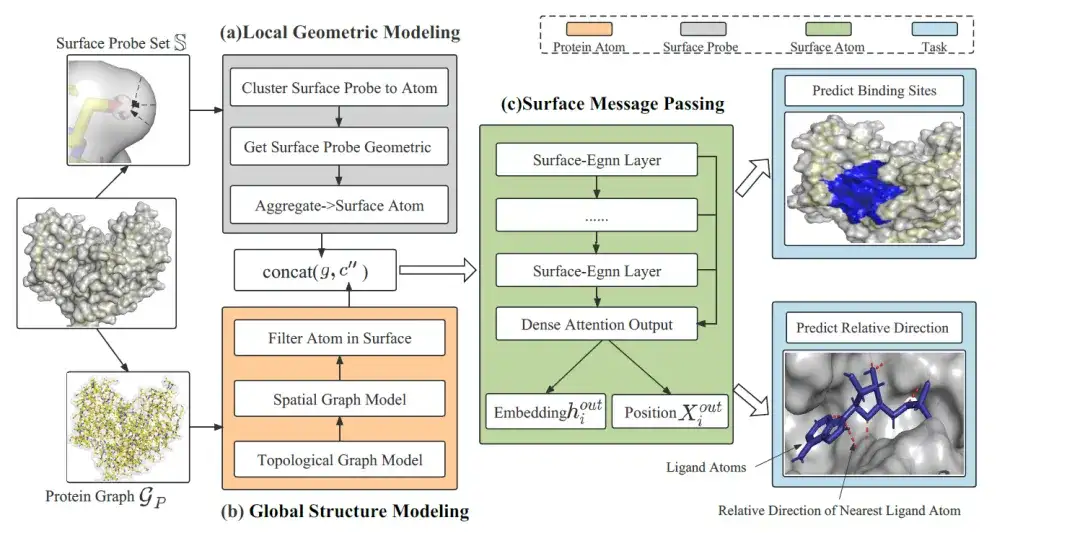
첫 번째 모듈은 각 표면 원자의 로컬 기하학적 정보를 추출하는 데 사용되는 로컬 기하학적 모델링 모듈입니다. 두 번째 모듈은 단백질의 화학적, 공간적 구조를 설명하는 데 사용되는 전역 구조 모델링 모듈입니다. 마지막 모듈은 표면 메시지 전달 모듈로, 표면 원자에 대한 등가 정보를 전송하여 표면 기하학을 포착합니다.
로컬 지오메트리 모델링 모듈
각 단백질 원자의 국소적 기하학은 근처 영역이 결합 부위의 일부가 되기에 적합한지 여부를 결정합니다.
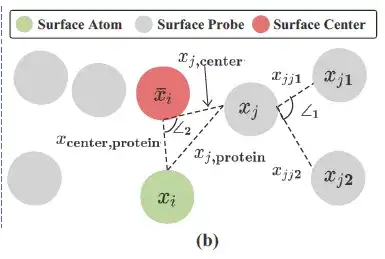
위 그림에서 보듯이, 연구자들은 각 단백질 표면 원자(위 그림에서 녹색으로 표시된 표면 원자) 주변에 표면 프로브(위 그림에서 회색으로 표시된 부분)를 사용하여 국소적인 기하학적 정보를 설명했습니다. 구체적으로, 각 표면 원자 i ∈ VS에 대해 그 주변의 표면 프로브는 S의 하위 집합에 의해 반환됩니다. 즉, 다음과 같습니다.
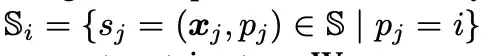
연구진은 Si를 기반으로 기하학적 정보를 구축하고 Si의 모든 3D 좌표의 중심/평균값(위 그림의 빨간색, 표면 중심)을 xi¯로 기록했습니다.
글로벌 구조 모델링 모듈
결합 부위는 주로 표면 원자로 구성되지만, 단백질의 전체 구조는 리간드 상호작용뿐만 아니라 결합 부위 형성에도 영향을 미치므로 모델링이 필요합니다.
연구자들은 화학 그래프 모델링과 공간 그래프 모델링이라는 두 가지 연결된 프로세스를 통해 이 목표를 달성했습니다. 그 결과 생성된 글로벌 구조 모델링 모듈은 원자 유형, 화학 결합, 상대적 공간 위치 등을 포함한 전체 단백질에 대한 정보를 처리하는 역할을 합니다.
표면 정보 전송 모듈
표면 원자의 국소적 기하학적 특징과 전역적 인코딩 특징이 주어지면, 이 모듈은 표면 맵에 대한 등가 정보 전송을 수행하여 단백질 표면 원자의 모든 특징을 업데이트합니다.
연구 결과: EquiPocket은 기준 모델과 비교하여 10-20%만큼 성능을 향상시킵니다.
실험에서 연구자들은 EquiPocket을 다음 기준 모델과 비교하기로 했습니다.
* 기하학 기반 방법: Fpocket
* 머신러닝 방식 : P2rank
* CNN 기반 방법: DeepSite, Kalasanty, DeepSurf, RecurPocket
* 토폴로지 기반 모델: GAT, GCN 및 GCN2
* 공간 그래프 기반 모델: SchNet, EGNN
모델을 평가하는 데 사용된 측정 항목에는 DCC(예측 결합 부위 중심과 실제 결합 부위 중심 사이의 거리), DCA(예측 결합 부위 중심과 리간드 그리드 사이의 최단 거리), 실패율(예측 결합 부위 중심이 없는 샘플링 속도)이 포함됩니다. 아래 표는 COACH 420, HOLO4K 및 PDBbind에 대한 결합 사이트 예측 결과를 보여줍니다.
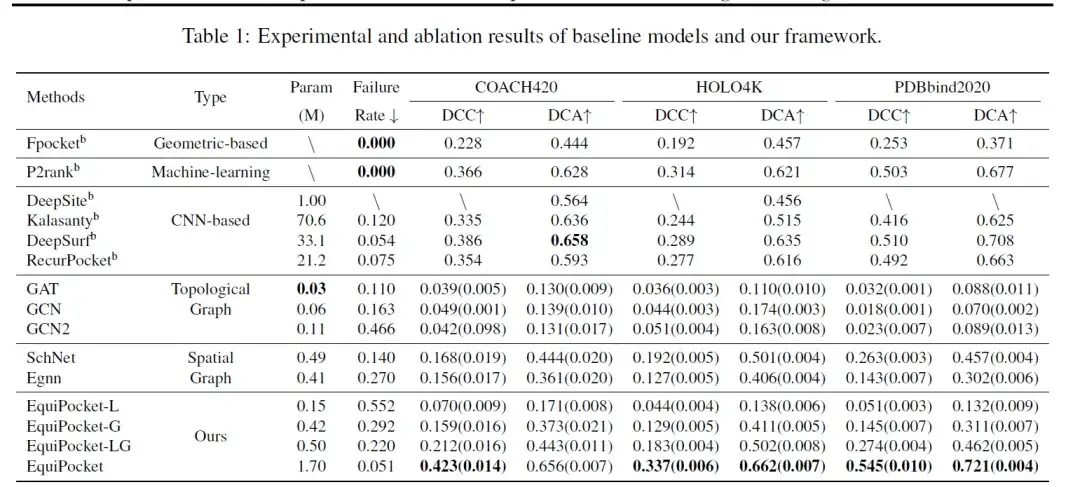
표의 데이터에서 볼 수 있듯이,기하학 기반 방법인 Fpocket은 성능이 좋지 않습니다.이 방법은 단백질의 기하학적 특징만을 사용하므로 실패율은 0입니다. 머신러닝 방법인 P2rank는 랜덤 포레스트와 단백질 표면의 기하학적 정보를 결합하여 성능을 크게 향상시켰습니다.
CNN 기반 방법(DeepSite, Kalasanty, DeepSurf, RecurPocket)은 기하 기반 방법보다 훨씬 더 나은 성능을 보입니다.그 중 DCC와 DCA는 50%보다 개선이 크지만, 많은 수의 매개변수와 컴퓨팅 리소스가 필요합니다. 그 중, 앞서 제안된 방법인 DeepSite와 Kalasanty는 단백질 크기의 변화에 제약을 받으며, 대형 단백질을 처리하는 능력이 부족하여 예측에 실패할 가능성이 있습니다.
그래프 모델의 경우,위상 그래프 모델(GCN, GAT, GCN2)의 성능이 좋지 않습니다.이는 주로 원자 및 화학 결합 정보만 고려하고 단백질의 공간적 구조는 무시하기 때문입니다. 공간 그래프 모델(SchNet, EGNN)의 성능은 일반적으로 위상 그래프 모델의 성능보다 우수합니다. EGNN은 원자의 속성과 상대적/절대적 공간 위치를 사용하는데, 이 방법이 더 효과적입니다. SchNet은 원자의 상대적 거리에 따라서만 임베딩을 업데이트하지만, 공간 그래프 모델의 성능은 CNN 기반 및 기하 기반 방법보다 떨어집니다. 왜냐하면 전자는 충분한 기하학적 특징을 얻을 수 없고 단백질 크기 변화 문제를 해결할 수 없기 때문입니다.
위의 결과는 다음을 보여줍니다.단백질 표면의 기하학적 정보와 다단계 구조 정보는 결합 부위 예측에 매우 중요합니다.
또한 이는 현재 GNN 모델의 한계를 반영하는 것으로, 즉 단백질 표면에서 충분한 기하학적 정보를 수집하기 어렵거나 필요한 컴퓨팅 리소스가 너무 커서 단백질과 같은 거대 분자 시스템에 적용하기 어렵다는 것입니다. 따라서 EquiPocket 프레임워크는 원자 수준에서 화학적, 공간적 정보를 업데이트할 수 있을 뿐만 아니라 과도한 계산 리소스를 요구하지 않고도 기하학적 정보를 효율적으로 수집할 수 있습니다.이번 연구의 성과는 이전 결과보다 10-20%만큼 향상되었습니다.
작은 분자 리간드부터 생물학적 거대 분자까지 AI가 단백질 구조를 심층적으로 해석합니다.
모든 식물, 동물, 인간의 세포 안에는 단백질, 핵산, 당과 같은 분자로 구성된 수십억 개의 분자 기계가 있으며, 그 중 어떤 부분도 단독으로 기능할 수 없습니다. 수백만 가지의 조합으로 이러한 분자 기계가 어떻게 상호 작용하는지 이해해야만 생명에 대한 더 깊은 이해를 얻을 수 있습니다.
올해 5월, 구글 딥마인드는 단백질, 핵산, 소분자, 이온, 변형 잔류물 등의 복합체에 대한 공동 구조 예측을 수행할 수 있는 AlphaFold3 모델을 출시했습니다. 단백질과 소분자 리간드 사이의 상호작용은 약물 작용 메커니즘의 핵심입니다. AlphaFold3는 첨단 딥러닝 알고리즘을 통해 단백질-리간드 결합의 3차원 구조를 정확하게 예측할 수 있으며, 그 정확도는 기존 도킹 도구의 정확도를 훨씬 뛰어넘습니다.
신약개발 측면에서는AlphaFold3가 예측한 단백질-리간드 구조를 통해 연구자들은 새로운 약물 후보물질을 보다 효과적으로 선별하고 설계하고 약물 발견 프로세스를 가속화할 수 있습니다. 기존 약물을 최적화하는 측면에서 이 도구는 표적 단백질과의 결합 모드를 개선하여 기존 약물의 효능을 향상시키거나 부작용을 줄임으로써 기존 약물을 최적화하는 데에도 사용될 수 있습니다.
소분자 리간드 외에도,단백질도 생물학적 기능을 발휘하려면 DNA나 당과 같은 생물학적 거대 분자와 결합해야 합니다.현재 수천 개의 단백질 구조 복합체가 실험적 방법을 통해 단백질 데이터베이스에 저장되어 있습니다. 그러나 기존의 실험 방법은 시간이 많이 걸리고 비용도 많이 드는 반면, 머신 러닝을 기반으로 한 예측 방법은 이러한 과제를 쉽게 해결할 수 있습니다.
올해 2월, 난징 농업대학 연구팀은 "ULDNA: 고정확도 단백질-DNA 결합 부위 예측을 위해 LSTM-Attention 네트워크와 비지도 다중 소스 언어 모델 통합"이라는 제목의 연구 논문을 생물학 분야의 중요 학술지인 Briefings in Bioinformatics에 온라인 게재했습니다.단백질-DNA 결합 부위를 예측하기 위해 새로운 딥러닝 예측 방법인 ULDNA가 개발되었습니다.
서류 주소:
https://academic.oup.com/bib/article/25/2/bbae040/7606634

ULDNA의 핵심 아이디어는 단백질 언어 모델을 사용하여 시퀀스에 대한 특징 표현을 설계한 다음, 장단기 기억 네트워크(LSTM-Attention Network)를 주의 메커니즘과 결합하여 DNA 결합 부위 예측 모델을 훈련하는 것입니다. 연구진은 PDNA-128, PDNA-316, PDNA-335(단백질 서열 수는 40~600개)를 포함한 7개의 벤치마크 데이터 세트를 선택하고 ULDNA에 대한 포괄적인 테스트를 수행했습니다. 실험 결과는 다음과 같습니다.ULDNA는 모든 데이터 세트에서 좋은 성능을 보였으며, 예측 성능은 다른 9가지 주요 방법보다 훨씬 우수했습니다.
DNA 외에도 당은 모든 유기체의 세포 표면에 널리 존재하며, 렉틴, 항체, 효소, 운반체 등 다양한 단백질 계열과 상호 작용하여 면역 반응, 세포 분화, 신경 발달과 같은 핵심적인 생물학적 과정을 조절합니다.탄수화물과 단백질의 상호작용 메커니즘을 이해하는 것은 탄수화물 약물을 개발하는 기초입니다.그러나 탄수화물 구조의 다양성과 복잡성, 특히 단백질 결합 부위의 다양성과 복잡성은 실험 데이터 수집과 약물 설계에 어려움을 야기합니다.
최근 중국 과학 아카데미의 한 팀이 DeepGlycanSite라는 딥 러닝 모델을 개발했는데, 이 모델은 주어진 단백질 구조에서 당 결합 부위를 정확하게 예측할 수 있습니다. DeepGlycanSite는 단백질의 기하학적, 진화적 특징을 Transformer 아키텍처를 기반으로 한 심층적 등가 그래프 신경망으로 통합합니다.이 기술의 성능은 기존의 고급 방법을 크게 능가하며 다양한 당 분자의 결합 부위를 효과적으로 예측할 수 있습니다.
돌연변이 연구와 결합한 DeepGlycanSite는 중요한 G 단백질 결합 수용체에 대한 구아노신-5'-이중인산 당 인식 부위를 밝혀냈습니다. 이러한 연구 결과는 DeepGlycanSite가 당 결합 부위 예측에 유용하며 치료적으로 중요한 단백질의 탄수화물 조절에 대한 분자적 메커니즘에 대한 통찰력을 제공할 수 있음을 시사합니다.

"DeepGlycanSite를 이용한 고정확도 탄수화물 결합 부위 예측"이라는 제목의 이 연구는 2024년 6월 17일 Nature Communications에 게재되었습니다.
서류 주소:
https://www.nature.com/articles/s41467-024-49516-2
요약하자면, 단백질은 생물체의 중요한 분자이며 세포의 구조와 기능에 중요한 역할을 합니다. 단백질의 구조를 연구하는 것은 생명 과정을 이해하고, 질병 메커니즘을 밝히고, 약물을 개발하는 데 매우 중요합니다. 오늘날 머신러닝은 과학자들에게 생명의 신비를 이해하는 새로운 문을 열어주고 있습니다.
참고문헌:
1.https://openreview.net/forum?id=1vGN3CSxVs
2.https://mp.weixin.qq.com/s/aGzcr0ncQA-jBy-vTGC35Q
3.https://www.jiqizhixin.com/articles/2024-05-09
4.https://news.njau.edu.cn/2024/0