Command Palette
Search for a command to run...
ICML, Tsinghua AIR 등이 공동으로 선정하여 기존 SOTA를 능가하는 단백질 언어 모델 ESM-AA를 출시

세포 내에서 수많은 생화학 반응을 주도하는 단백질은 세포라는 미세한 세계에서 건축가와 엔지니어의 역할을 합니다. 그들은 생명 활동을 촉진할 뿐만 아니라, 생물체의 형태와 기능을 구성하고 유지하는 기본 구성 요소 역할을 합니다. 생명의 거대한 청사진을 뒷받침하는 것은 바로 단백질 간의 상호작용과 상승효과입니다.
그러나 단백질의 구조는 복잡하고 변화하기 쉬우며, 전통적인 실험 방법으로 단백질 구조를 분석하는 것은 시간이 많이 걸리고 노력이 많이 듭니다. 단백질 언어 모델(PLM)이 등장했습니다. 심층학습 기술을 활용하여 대량의 단백질 서열 데이터를 분석하고, 단백질의 생화학적 법칙과 공진화 패턴을 학습함으로써 단백질 구조 예측, 적응성 예측 및 단백질 설계 분야에서 괄목할 만한 성과를 거두었으며, 단백질 공학의 발전을 크게 촉진시켰습니다.
PLM은 잔류물 규모에서 큰 성공을 거두었지만 원자 수준의 정보를 제공하는 능력에는 한계가 있습니다. 이에 대해 청화대 지능산업연구소 주하오(周浩) 연구원은 베이징대, 난징대, 슈이무 분자팀과 협력해다중 스케일 단백질 언어 모델 ESM-AA(ESM All Atom)가 제안되었습니다.잔류물 확장 및 다중 스케일 위치 인코딩과 같은 훈련 메커니즘을 설계함으로써 원자 스케일 정보를 처리하는 능력이 확장되었습니다.
타겟 리간드 결합과 같은 작업에서 ESM-AA의 성능은 크게 향상되어 ESM-2와 같은 현재의 SOTA 단백질 언어 모델을 능가하고 Uni-Mol과 같은 현재의 SOTA 분자 표현 학습 모델보다 뛰어납니다. 관련 연구는 "ESM All-Atom: Unified Molecular Modeling을 위한 다중 스케일 단백질 언어 모델"이라는 제목으로 출판되었습니다.최고의 머신러닝 컨퍼런스인 ICML에 게재되었습니다.

서류 주소:
https://icml.cc/virtual/2024/poster/35119
오픈소스 프로젝트인 "awesome-ai4s"는 100개가 넘는 AI4S 논문 해석을 모아 방대한 데이터 세트와 도구를 제공합니다.
https://github.com/hyperai/awesome-ai4s
데이터 세트: 단백질과 분자 데이터의 혼합 데이터 세트가 구성되었습니다.
사전 훈련 작업에서,이 연구에서는 원자 좌표와 같은 구조적 정보를 포함하는 단백질과 분자 데이터의 결합된 데이터 세트를 사용했습니다.
단백질 데이터 세트의 경우, 이 연구에서는 800만 개의 높은 신뢰도의 AlphaFold2 예측 단백질 서열과 구조가 포함된 AlphaFold DB를 사용했습니다.
분자 데이터 세트의 경우, 이 연구에서는 ETKDG와 MMFF 분자력장에서 생성된 데이터를 사용했는데, 여기에는 1,900만 개의 분자와 2억 900만 개의 구성이 포함되어 있습니다.
연구진은 ESM-AA를 훈련할 때 먼저 단백질 데이터 세트 Dp와 분자 데이터 세트 Dm을 최종 데이터 세트로 혼합했습니다. 즉, D=Dp∪Dm입니다. Dm의 분자의 경우 원자로만 구성되어 있으므로 코드 변환 시퀀스 X̄는 잔류물이 없는 모든 원자 Ā의 순서화된 집합입니다. 즉, R̄=∅입니다. 분자 데이터는 사전 학습에 사용되므로 ESM-AA는 단백질과 분자를 모두 입력으로 받을 수 있다는 점에 유의해야 합니다.
ESM-AA 모델 구축: 통합 분자 모델링을 달성하기 위한 다중 스케일 사전 학습 및 인코딩
다중 언어 코드 전환 방법에서 영감을 받은 ESM-AA는 예측 및 단백질 설계 작업을 수행할 때 먼저 일부 잔류물을 무작위로 압축 해제하여 다중 스케일 코드 전환 단백질 시퀀스를 생성합니다. 그런 다음 이러한 시퀀스는 신중하게 설계된 다중 스케일 위치 인코딩을 통해 훈련되며, 그 효과는 잔류물과 원자 스케일에서 입증되었습니다.
단백질 분자 작업, 즉 단백질과 소분자가 관련된 작업을 처리할 때 ESM-AA는 추가적인 모델 지원이 필요하지 않으며 사전 훈련된 모델의 기능을 최대한 활용할 수 있습니다.
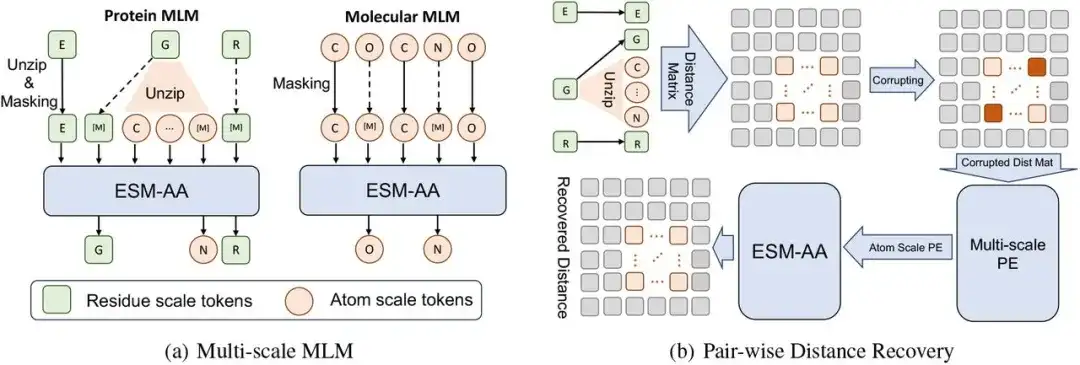
본 연구의 다중 스케일 사전 학습 프레임워크는 다중 스케일 마스크 언어 모델(MLM)과 쌍별 거리 복구로 구성됩니다.
구체적으로, 잔류물 규모에서 단백질 X는 L개의 잔류물의 시퀀스로 볼 수 있습니다. 즉, X = (r1,…,ri,…,rL). 각 잔류물 ri는 N개의 원자 A Ai={a1i,…,aNi}로 구성됩니다. 코드 전환 단백질 서열 X̅을 구성하기 위해, 우리는 잔류물 세트를 무작위로 선택하고 해당 원자를 X에 삽입하여 압축 해제 프로세스를 구현했습니다. 이 프로세스에서 연구자들은 압축 해제된 원자를 순서대로 배열했고, 마지막으로 원자 세트 Ai를 X에 삽입한 후(즉, 잔류물 ri를 압축 해제) 코드 전환 서열 X̄을 얻었습니다.
그 다음에,연구자들은 코드 전환 시퀀스 X̄에 대해 마스크 언어 모델링을 수행했습니다.
먼저, X̄의 일부 원자나 잔류물을 무작위로 마스크 처리하고 모델이 주변 맥락을 사용하여 원래 원자나 잔류물을 예측하도록 합니다. 연구자들은 또 다른 사전 훈련 과제로 쌍방향 거리 복구(PDR)를 사용했습니다. 즉, 원자 규모의 구조 정보는 좌표에 노이즈를 추가하여 파괴되고, 파괴된 원자 간 거리 정보는 모델 입력으로 사용되어 모델이 이들 원자 간의 정확한 유클리드 거리를 복원해야 합니다.
다양한 잔류물에 걸친 장거리 구조 정보와 단일 잔류물 내의 원자 규모 구조 정보 사이의 의미적 차이를 고려하여, 이 연구에서는 잔류물 내의 PDR만 계산하며, 이를 통해 ESM-AA가 다양한 잔류물 내의 다양한 구조 지식을 학습할 수 있습니다.
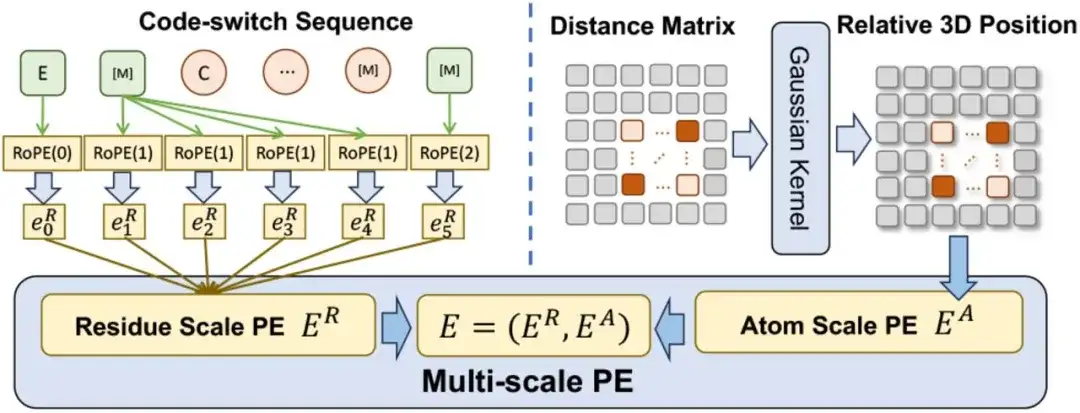
다중 스케일 위치 인코딩 측면에서, 연구자들은 코드 전환 시퀀스의 위치 관계를 인코딩하기 위해 다중 스케일 위치 인코딩 E를 설계했습니다. E에는 잔류 규모 위치 코드 ER과 원자 규모 위치 코드 EA가 포함되어 있습니다.
응급실의 경우,연구진은 기존 인코딩 방식을 확장하여 순수한 잔류물 서열을 다룰 때 원래 인코딩과의 일관성을 유지하면서 잔류물-원자 관계를 인코딩할 수 있도록 했습니다.EA의 경우,원자 간의 관계를 파악하기 위해 이 연구에서는 공간 거리 행렬을 직접 사용하여 3차원 위치를 인코딩했습니다.
다중 스케일 인코딩 방식은 사전 학습이 모호한 위치 관계의 영향을 받지 않도록 보장하므로 ESM-AA가 두 스케일 모두에서 효과적으로 작동할 수 있다는 점이 언급할 가치가 있습니다.
다중 스케일 PE를 Transformer에 통합할 때, 이 연구는 먼저 Transformer의 사인파 코딩을 잔여 스케일 위치 코딩 ER로 대체하고, 원자 스케일 위치 코딩 EA를 자기 주의 계층의 바이어스 항으로 간주했습니다.
연구 결과: 단백질 이해 최적화를 위한 분자 지식 통합
다중 스케일 통합 사전 학습 모델의 효과를 검증하기 위해, 이 연구에서는 단백질과 소분자가 관련된 다양한 작업에서 ESM-AA의 성능을 평가했습니다.
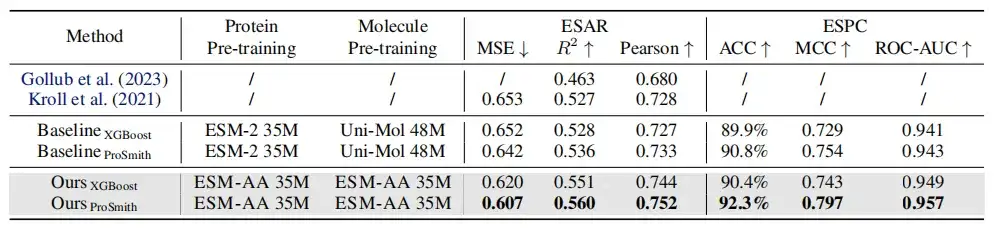
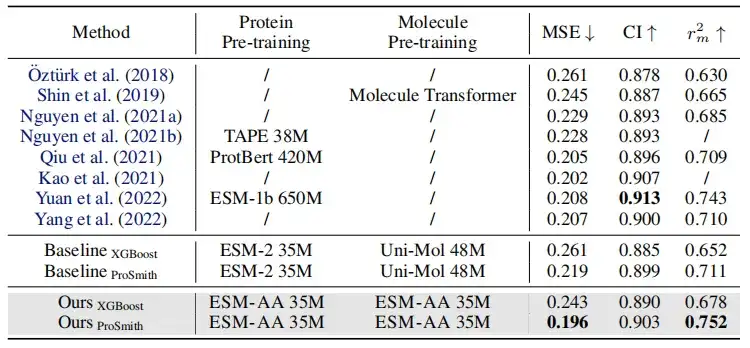
위 표에서 보듯이 효소-기질 친화도 회귀 과제, 효소-기질 쌍 분류 과제, 약물-표적 친화도 회귀 과제의 성능 비교에서,대부분의 측정 기준에서 ESM-AA는 다른 모델보다 우수한 성과를 보이며 최첨단 결과를 달성합니다.또한 ESM-AA를 기반으로 한 미세 조정 전략(ProSmith 및 XGBoost 등)은 두 개의 독립적인 분자 사전 훈련 모델과 단백질 사전 훈련 모델을 결합한 버전보다 지속적으로 우수한 성과를 보였습니다(표 1 및 2의 마지막 네 행 참조).
주목할 점은 다음과 같습니다.ESM-AA는 더 큰 매개변수 크기를 가진 사전 학습된 모델을 사용하는 방법보다 더 우수할 수도 있습니다.(예를 들어, 표 2의 5번째, 7번째, 마지막 행을 비교하는 경우).
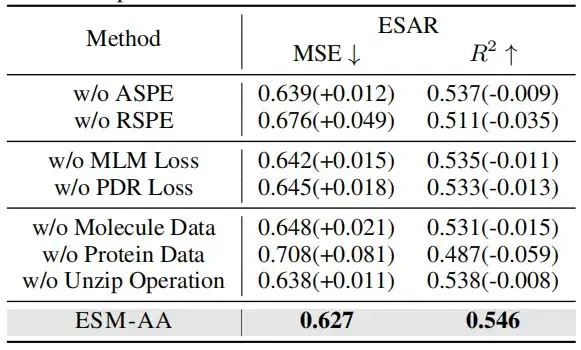
다중 스케일 위치 인코딩의 효과를 검증하기 위해 본 연구에서는 두 가지 경우에 대한 절제 실험을 수행했습니다. 하나는 원자 스케일 위치 인코딩(ASPE)을 사용하지 않는 경우입니다. 다른 하나는 유전적 규모 위치 인코딩(RSPE)을 사용하지 않는 것입니다.
분자나 단백질 데이터를 제거하면 모델 성능이 크게 떨어졌습니다. 흥미로운 점은 단백질 데이터를 제거함으로써 발생하는 성능 저하는 분자 데이터를 제거함으로써 발생하는 성능 저하보다 더 명백하다는 것입니다. 이는 모델이 단백질 데이터로 훈련되지 않았을 경우 단백질 관련 지식을 빠르게 잃어버려 전반적인 성능이 크게 저하된다는 것을 의미합니다. 하지만,분자 데이터가 없더라도 이 모델은 감압 작업을 통해 원자 수준의 정보를 얻을 수 있습니다.
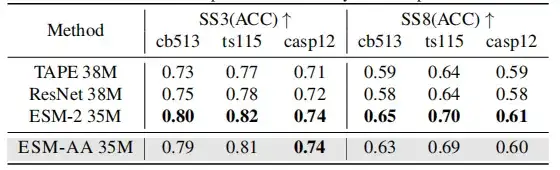
ESM-AA는 기존 PLM을 기반으로 개발되었으므로, 이 연구에서는 ESM-AA가 여전히 단백질에 대한 포괄적 이해를 유지하는지 확인하고, 2차 구조 예측 및 비지도 접촉 예측 작업을 사용하여 단백질 구조를 이해하는 단백질 사전 훈련 모델의 능력을 테스트하고자 합니다.
결과는 ESM-AA가 이 유형의 연구에서 최적의 성능을 달성하지 못할 수 있지만그러나 2차 구조 예측 및 접촉 예측 성능은 ESM-2와 유사합니다.
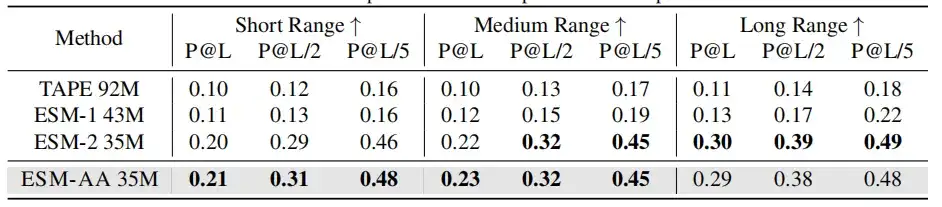
분자 벤치마킹에서ESM-AA는 대부분의 작업에서 Uni-Mol과 비슷한 성능을 보입니다.이 방법은 여러 경우에서 여러 분자 특정 모델보다 우수한 성능을 보이며, 분자 작업을 위한 강력한 접근 방식이라는 잠재력을 보여줍니다.
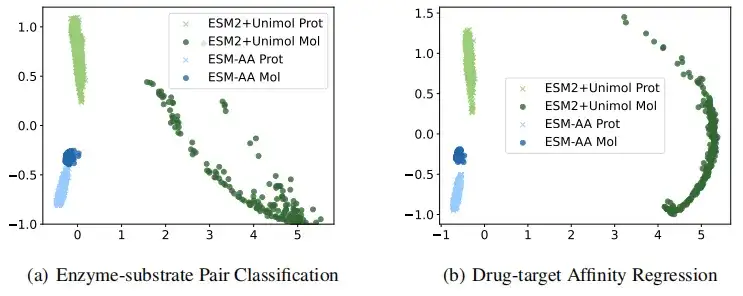
ESM-AA가 더 높은 품질의 단백질과 소분자 표현을 얻는다는 것을 보다 직관적으로 보여주기 위해, 이 연구에서는 효소-기질 쌍 분류와 약물 표적 친화도 회귀 작업에서 ESM-AA와 ESM-2+Uni-Mol이 추출한 표현을 시각적으로 비교했습니다. 결과는 다음과 같습니다ESM-AA 모델은 단백질과 분자 데이터 모두에 대해 더욱 응집력 있는 의미적 표현을 생성할 수 있어 두 개의 별도 사전 학습된 모델보다 성능이 뛰어납니다.
단백질 언어 모델, 대규모 언어 모델의 다음 단계
1970년대 이래로 점점 더 많은 과학자들이 "21세기는 생물학의 세기"라고 믿게 되었습니다. 작년 7월, 포브스는 LLM이 생물학 분야의 새로운 변화의 선두에 사람들을 배치할 것이라고 가정한 긴 기사를 썼습니다. 생물학은 해독, 프로그래밍이 가능하고, 어떤 면에서는 디지털화도 가능한 시스템으로 밝혀졌습니다.LLM은 자연어를 제어하는 놀라운 능력을 갖추고 있어 생물학적 언어를 해독할 수 있는 잠재력을 제공합니다.이로 인해 단백질 언어 모델은 이 시대의 가장 인기 있는 분야 중 하나가 되었습니다.
단백질 언어 모델은 생물학에서 AI 기술을 최첨단으로 적용한 사례입니다. 단백질 서열의 패턴과 구조를 학습함으로써 단백질의 기능과 형태를 예측할 수 있는데, 이는 신약 개발, 질병 치료 및 기초 생물학 연구에 매우 중요합니다.
이전에는 ESM-2 및 ESMFold와 같은 단백질 언어 모델이 AlphaFold와 비슷한 정확도를 보였으며, "고아 단백질"에 대한 처리 속도가 더 빠르고 예측 기능이 더 정확했습니다. 이는 단백질 구조 예측 속도를 높일 뿐만 아니라 단백질 공학을 위한 새로운 도구를 제공하여 연구자들이 특정 기능을 갖춘 완전히 새로운 단백질 서열을 설계할 수 있게 해줍니다.
더욱이 단백질 언어 모델의 개발은 소위 "스케일링 법칙"으로부터 이익을 얻습니다.즉, 모델 규모, 데이터 세트 크기 및 계산 노력이 증가함에 따라 모델 성능이 크게 향상됩니다.즉, 모델 매개변수가 증가하고 학습 데이터가 축적됨에 따라 단백질 언어 모델의 역량이 질적으로 비약적으로 향상될 것입니다.
지난 2년 동안 단백질 언어 모델도 비즈니스 커뮤니티에서 급속한 발전 단계에 접어들었습니다. 2023년 7월, 바이두 바이오사이언스와 청화대학교는 매개변수 크기가 최대 1000억(100B)인 xTrimo 단백질 일반 언어 모델(xTrimoPGLM)이라는 모델을 공동으로 제안했습니다. 이 모델은 여러 단백질 이해 작업에서 다른 고급 기준 모델보다 상당히 우수한 성능을 보였습니다(15개 작업 중 13개).생성 작업에서 xTrimoPGLM은 자연 단백질 구조와 유사한 새로운 단백질 서열을 생성할 수 있습니다.
2024년 6월, AI 단백질 회사 Tushen Zhihe는 다음과 같이 발표했습니다.중국 최초의 자연어 단백질 모델인 TourSynbio™가 모든 연구자와 개발자에게 오픈 소스로 공개되었습니다.이 모델은 단백질 특성, 기능 예측, 단백질 설계 등의 기능을 포함하여 대화형 방식으로 단백질 문헌에 대한 이해를 달성합니다. 단백질 평가 데이터 세트를 비교하는 평가 지표 측면에서 GPT4를 능가하여 업계 최초가 되었습니다.
또한 ESM-AA가 대표하는 기술 연구의 획기적인 발전은 기술 발전이 '라이트 형제의 순간'을 지나 도약의 시대로 접어들고 있음을 의미할 수도 있습니다. 동시에 단백질 언어 모델의 적용은 의학 및 생물제약 분야에만 국한되지 않고 농업, 산업, 재료 과학 및 환경 복원 등 다양한 분야로 확장되어 이러한 분야의 기술 혁신을 촉진하고 인류에게 전례 없는 변화를 가져올 것입니다.