Command Palette
Search for a command to run...
Tutoriel Sur Le Séquençage Du Transcriptome Monocellulaire À Partir d'un Seul Échantillon : Contrôle Qualité, Clustering, Affichage (différentiel) Des Gènes
Date
Taille
2.42 GB
URL du document
Tutoriel sur l'analyse du séquençage du transcriptome d'une cellule unique
Les données utilisées dans cette étude proviennent d'un article publié dans Nature Medicine en juillet 2024. Vaccin néoantigène personnalisé et pembrolizumab dans le carcinome hépatocellulaire avancé : un essai de phase 1/2 Données de séquençage unicellulaire des cellules mononucléaires du sang périphérique du patient 6 (Yarchoan M et al. Nat Med. 2024;30:1044–1053.).
Introduction au séquençage de cellules uniques
Le séquençage unicellulaire est une biotechnologie sophistiquée qui permet aux scientifiques d’analyser en profondeur l’expression des gènes au niveau des cellules individuelles. Le processus commence généralement par un échantillon de tissu qui est d’abord dissocié en cellules individuelles pour former une suspension unicellulaire. Ensuite, en utilisant la technique eau dans huile, chaque cellule a été encapsulée individuellement pour garantir qu’elles restent indépendantes lors des analyses ultérieures.
Dans le séquençage unicellulaire, la molécule d'ARNm de chaque cellule est marquée avec un identifiant moléculaire unique, un processus appelé marquage UMI (Unique Molecular Identifier). Dans le même temps, chaque cellule se verra attribuer une même étiquette de code-barres, qui est utilisée pour suivre la source de chaque cellule pendant l'étape d'analyse des données. De cette façon, les chercheurs peuvent mesurer avec précision les informations de l’ARNm dans chaque cellule, comprenant ainsi l’état d’expression génétique de chaque cellule.
1. Fonction code-barres : enregistrement des informations sur les cellules
2. Application de l'UMI au séquençage unicellulaire : en fournissant un identifiant unique pour chaque molécule d'ARNm, il améliore non seulement la précision de la mesure de l'expression génétique, mais réduit également les erreurs qui peuvent survenir lors de l'amplification par PCR, améliorant ainsi la fiabilité et l'efficacité de l'ensemble du processus de séquençage.
Processus de base
Le processus de séquençage d'ARN unicellulaire (scRNA-seq) comprend principalement l'isolement et la capture de cellules individuelles, la lyse cellulaire, la transcription inverse (conversion de l'ARN en ADNc), l'amplification de l'ADNc et la préparation de la bibliothèque.
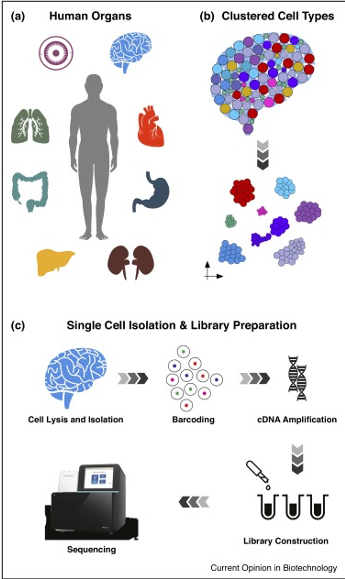
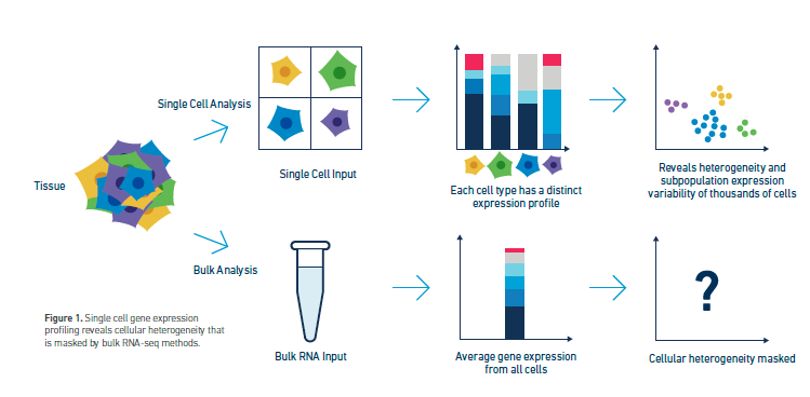
Source de l’image : Kulkarni, A., et al. (2019). «Au-delà du volume : une revue des méthodes et applications de transcriptomique à cellule unique« Curr Opin Biotechnol 58 : 129-136
Étapes de course
Nous allons maintenant entrer dans la partie pratique du tutoriel.
Démarrez le conteneur et téléchargez les données
1. Démarrez le conteneur
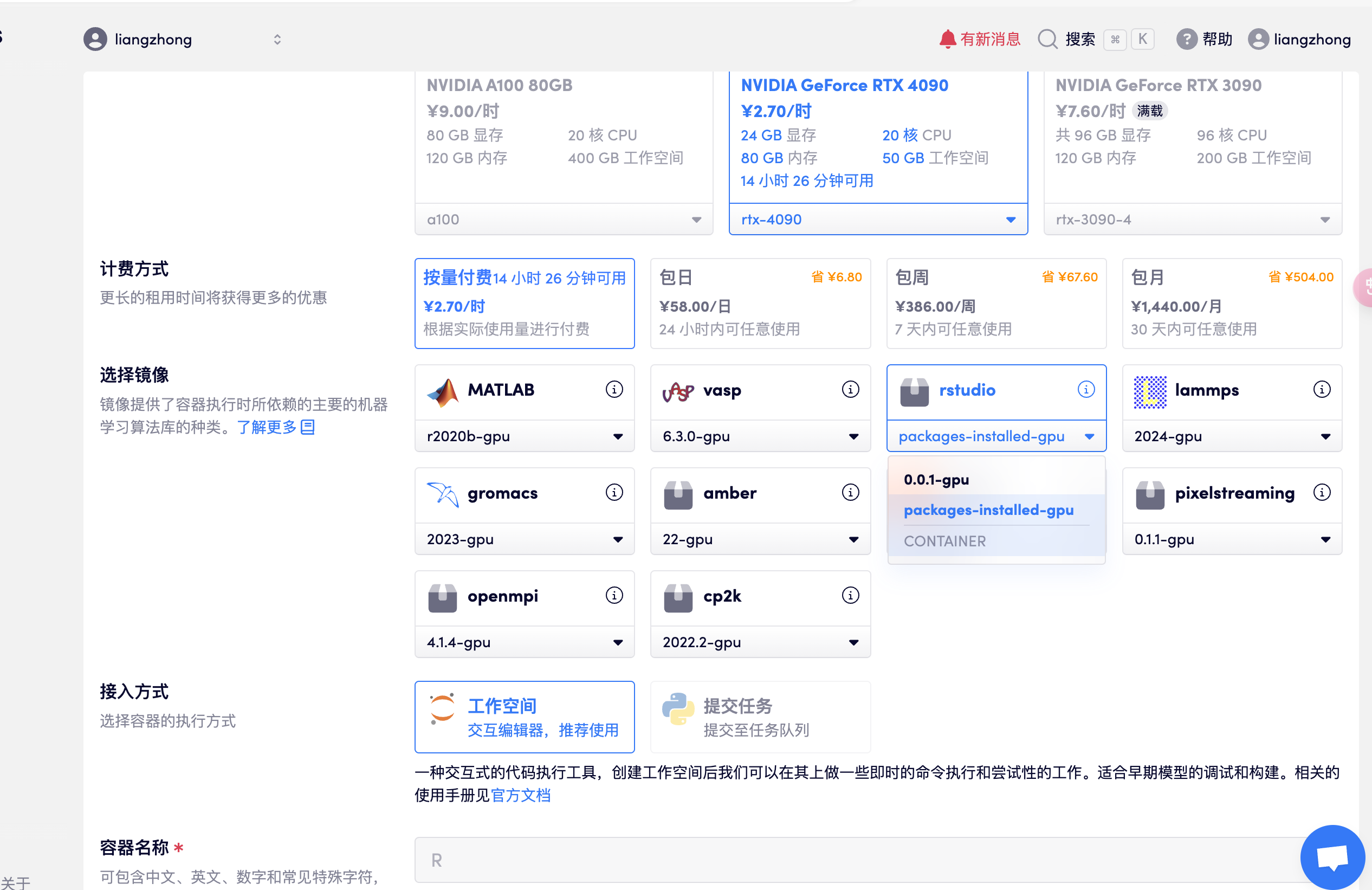
Créez un conteneur, ouvrez rstudio, sélectionnez la version package-installed-gpu et ouvrez directement l'adresse API pour accéder à R studio une fois les ressources allouées (cette étape nécessite une authentification par nom réel), comme indiqué dans la figure suivante :
- Remarque : après avoir accédé au logiciel, le compte et le mot de passe sont : rstudio
2. Télécharger les données
Téléchargez les données préparées à l'avance (les données peuvent être téléchargées et utilisées dans le dossier de données du répertoire de travail) :
(1) Données brutes de cellules individuelles séquencées par Illumina NextSeq 500 : barcodes.tsv.gz, features.tsv.gz, matrix.mtx.gz
(2) Données de référence pour l'annotation des cellules : pbmc_multimodal.h5seurat, ref_Human_all.RData
#存放的目录如下
#此处可新建自定义文件夹,指定输入输出文件目录,本教程以 output 为例
rstudio@****(用户名)-bs5z6fbf81vrmain:~/home/output/data$ ls
barcodes.tsv.gz features.tsv.gz matrix.mtx.gz
rstudio@****(用户名)-bs5z6fbf81vr-main:~/home/output/reference_annotation$ ls
pbmc_multimodal.h5seurat ref_Human_all.RDataEnsuite, entrez le code dans R Studio étape par étape selon les étapes du didacticiel.
#报错改为英文
Sys.setenv(LANGUAGE = "en")
options(stringsAsFactors = FALSE)
#清空所有数据
rm(list=ls())
set.seed(100)#!!!设置随机数,保证后续实验的可重复性1. Configurer l'objet Seurat
# I. Setup -----------------------------------------------------------------------
# ## a. Load libraries ----------------------------------------------------------------------
```{r libraries}
library(Seurat)
library(SeuratObject)
library(sctransform)
library(patchwork)
library(Seurat)
library(SeuratData)
library(patchwork)
library(SeuratData)
library(ggplot2)
library(tidyverse)
library(glmGamPoi)
library(tools)
library(dplyr)
library(patchwork)2. Chargement des données
# II. Load data-----------------------------------------------------------------------
### a. Load data-----------------------------------------------------------------
getwd()#查看当前工作目录
setwd("~/home/output/results")
#修改工作目录,此处可根据自定义文件夹,指定输入输出文件目录,本教程以 output 为例
pbmc.data <- Read10X(data.dir ="~/home/output/data")#设置 10x 原始数据的工作目录,读取
seurat <- CreateSeuratObject(counts = pbmc.data, project = "pbmc", min.cells = 3, min.features = 200)#创建 seurat 对象
seurat
An object of class Seurat
16356 features across 10066 samples within 1 assay
Active assay: RNA (16356 features, 0 variable features)
1 layer present: counts
#该样本有 10066 个细胞 ,10066 个单细胞的 RNA 表达数据,测序了 16356 个基因
#counts: 通常表示原始的基因表达计数数据
colnames([email protected])
[1] "orig.ident" "nCount_RNA" "nFeature_RNA" "percent.mt" "RNA_snn_res.0.1" "seurat_clusters"
[7] "celltype"
Les données pbmc.data sont présentées dans la figure ci-dessous, avec des lectures, les noms de lignes sont des gènes et les noms de colonnes sont des codes-barres de cellules.
pbmc.data[c("CD3D", "CD8A", "PTPRC"), 1:30] Matrice creuse 3 x 30 de classe « dgCMatrix » [[ suppression des noms de colonnes 30 « AAACCTGAGCGAAGGG-1 », « AAACCTGAGGCCCCTTG-1 », « AAACCTGAGGGTTTCT-1 » … ]]
CD3D 2 . 3 6 4 1 1 3 . 6 3 4 2 1 2 1 . 2 2 5 2 3 . 6 . 4 3 CD8A . 6 . . . 6 PTPRC 1 2 2 2 3 5 3 3 3 1 3 . 2 4 1 . 1 . 2 9 . 3 4 . 1 2 . 2 1 4
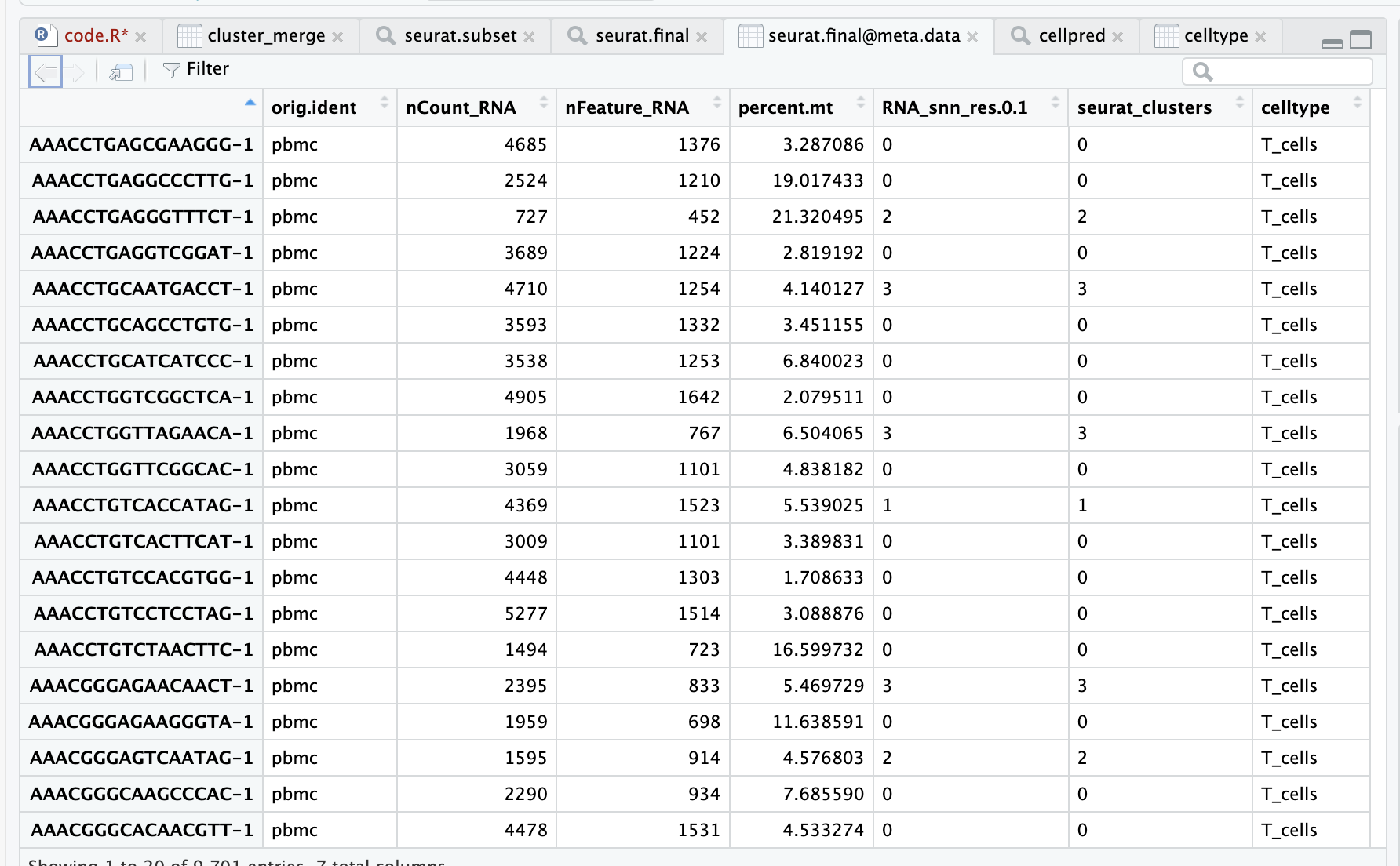
- Description des informations du tableau :
- Le nom de la ligne est codes-barres
- orig.ident est l'exemple d'informations,
- nCount_RNA est l'expression totale des gènes
- nFeature_RNA est le nombre de gènes
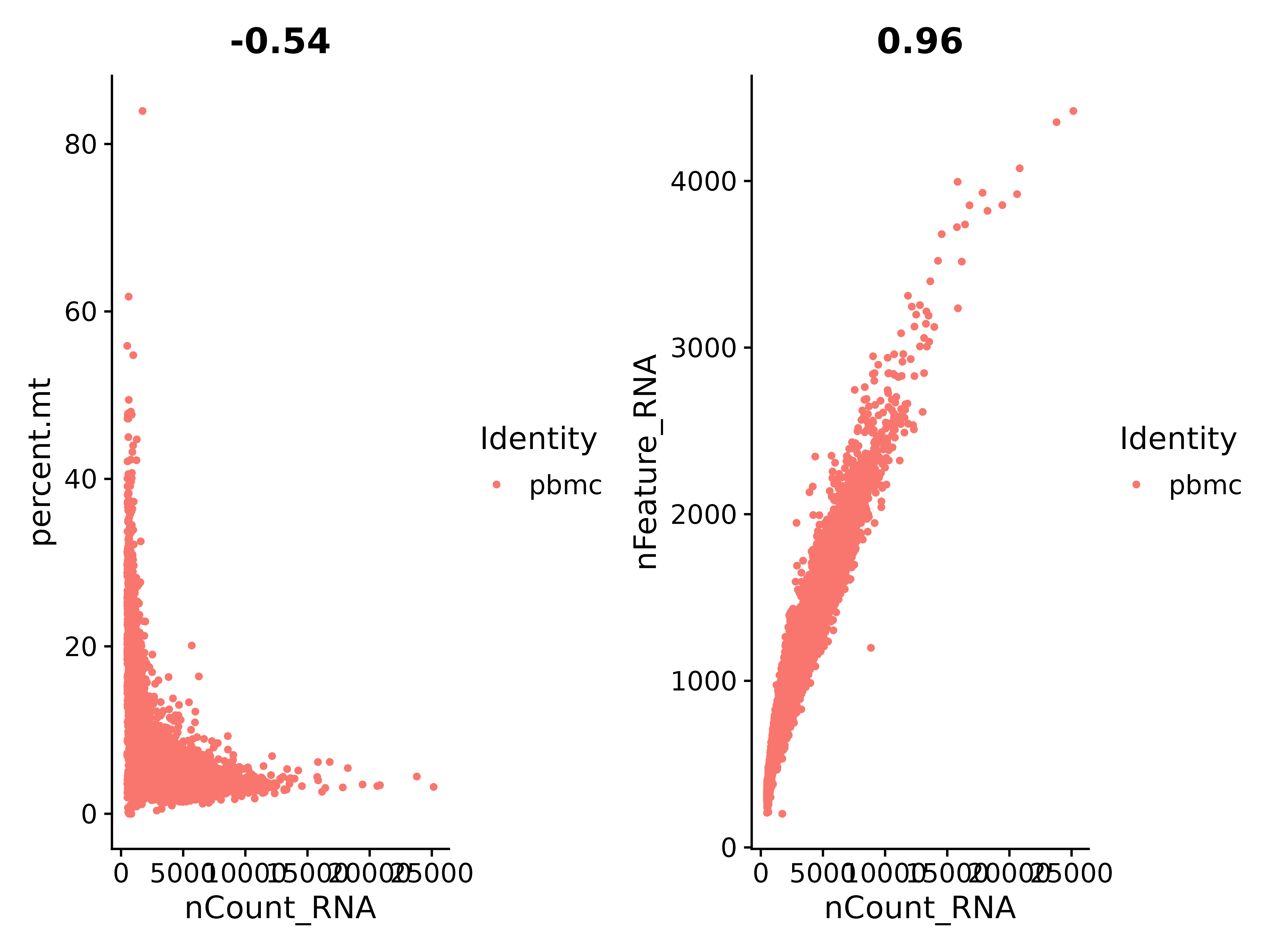
D'après la figure ci-dessus, vérifions le rapport entre nCount_RNA et percent.mt (gène mitochondrial) :
On peut voir que nCount_RNA et nFeature_RNA sont en proportion positive, avec un coefficient de 0,96, et la corrélation est très bonne.
Conseils : Tous les gènes commençant par MT- sont des gènes mitochondriaux.
3. Prétraitement standard
Les étapes suivantes couvrent le flux de travail de prétraitement standard pour les données scRNA-seq dans Seurat. Ces processus incluent la sélection et le filtrage des cellules en fonction des mesures de contrôle qualité, la normalisation et la mise à l’échelle des données, ainsi que la détection de caractéristiques très variables.
Seurat peut afficher les mesures de contrôle qualité et filtrer les cellules en fonction de n'importe quel critère défini par l'utilisateur.
# III. Pre-processing----------------------------------------------------------------
### a. Plots for percent mito, nFeature---------------------------------------------
```{r preprocessing}
# The [[ operator can add columns to object metadata. This is a great place to stash QC stats
seurat <-PercentageFeatureSet(seurat, pattern = "^MT-", col.name = "percent.mt")
# Use FeatureScatter is typically used to visualize feature-feature relationships, but can be used
# for anything calculated by the object, i.e. columns in object metadata, PC scores etc.
plot1 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.mt",)
plot2 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
plot1 + plot2
ggsave("质控_1.png", plot1 + plot2, width = 8, height = 6, dpi=600)
#plot3 <- FeatureScatter(seurat, feature1 = "nCount_RNA", feature2 = "percent.ribo")
#plot3
Ensuite, nous visualisons les mesures de contrôle qualité et les utilisons pour filtrer les cellules.
Regardez la proportion de gènes mitochondriaux et utilisez des graphiques en violon pour visualiser les indicateurs de contrôle qualité
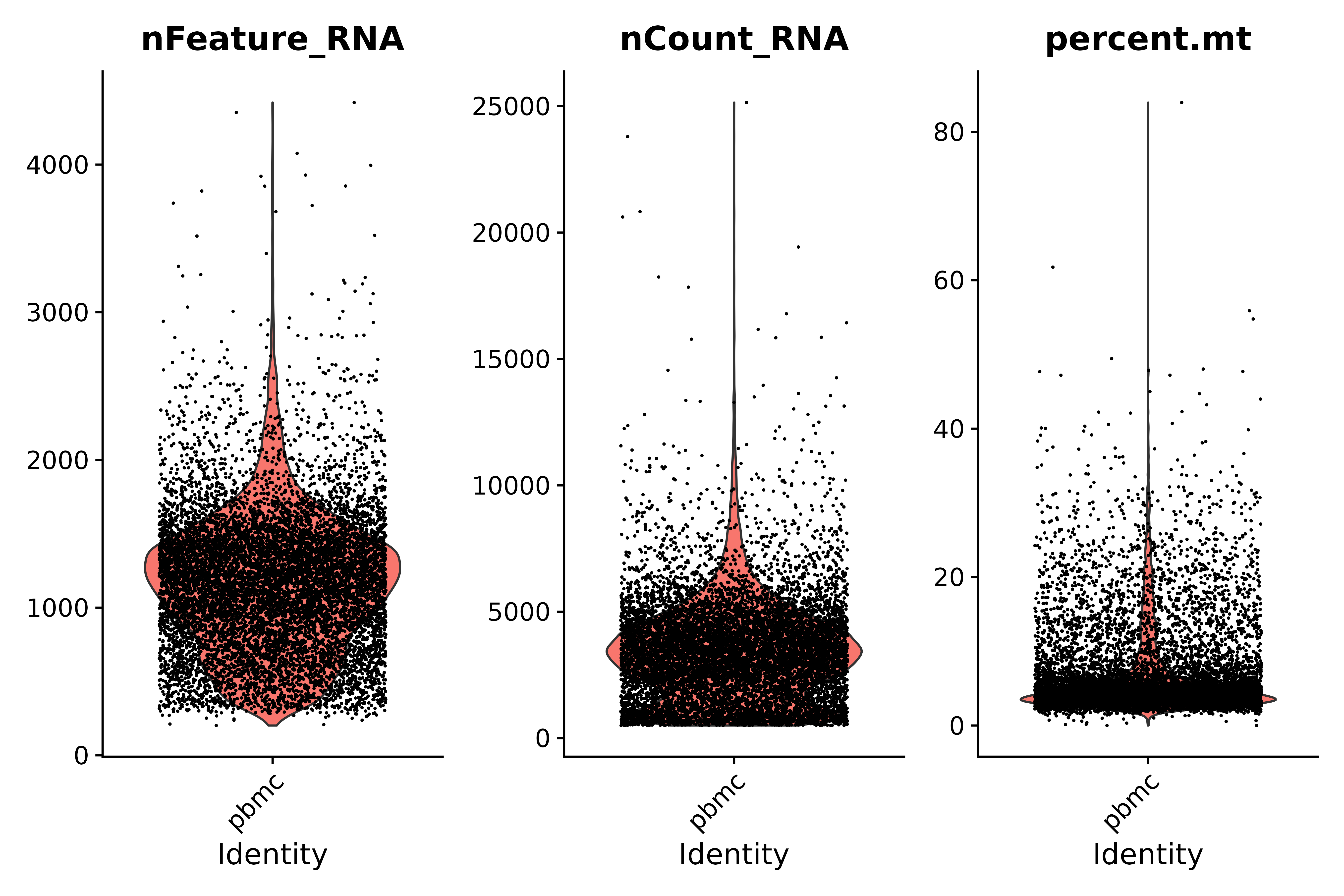
- Instructions de contrôle de qualité :
- Les cellules de faible qualité ou les gouttelettes vides ont généralement très peu de gènes, avec nFeature_RNA > 200 et nCount_RNA > 500.
- Les cellules diploïdes ou polyploïdes peuvent présenter un nombre anormalement élevé de gènes ; selon notre graphique, nFeature_RNA < 2500 et nCount_RNA > 500
- Les cellules de faible qualité/mourantes présentent généralement une hyperplasie mitochondriale étendue, pourcent.mt < 25
# Visualize QC metrics as a violin plot
vln1 <- VlnPlot(seurat, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"#,"percent.ribo"
),ncol=3)
vln1
ggsave("质控_2.png", vln1, width = 9, height = 6, dpi=600)seurat.subset <- subset(seurat, subset = nFeature_RNA > 200 & nFeature_RNA < 2500 & nCount_RNA > 500 & nCount_RNA < 10000 &percent.mt < 25)
vln3 <- VlnPlot(seurat.subset, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
vln3
ggsave("质控_3.png", vln3, width = 9, height = 6, dpi=600)
pdf("qc_plots.pdf");plot1 + plot2;vln1;vln3;dev.off()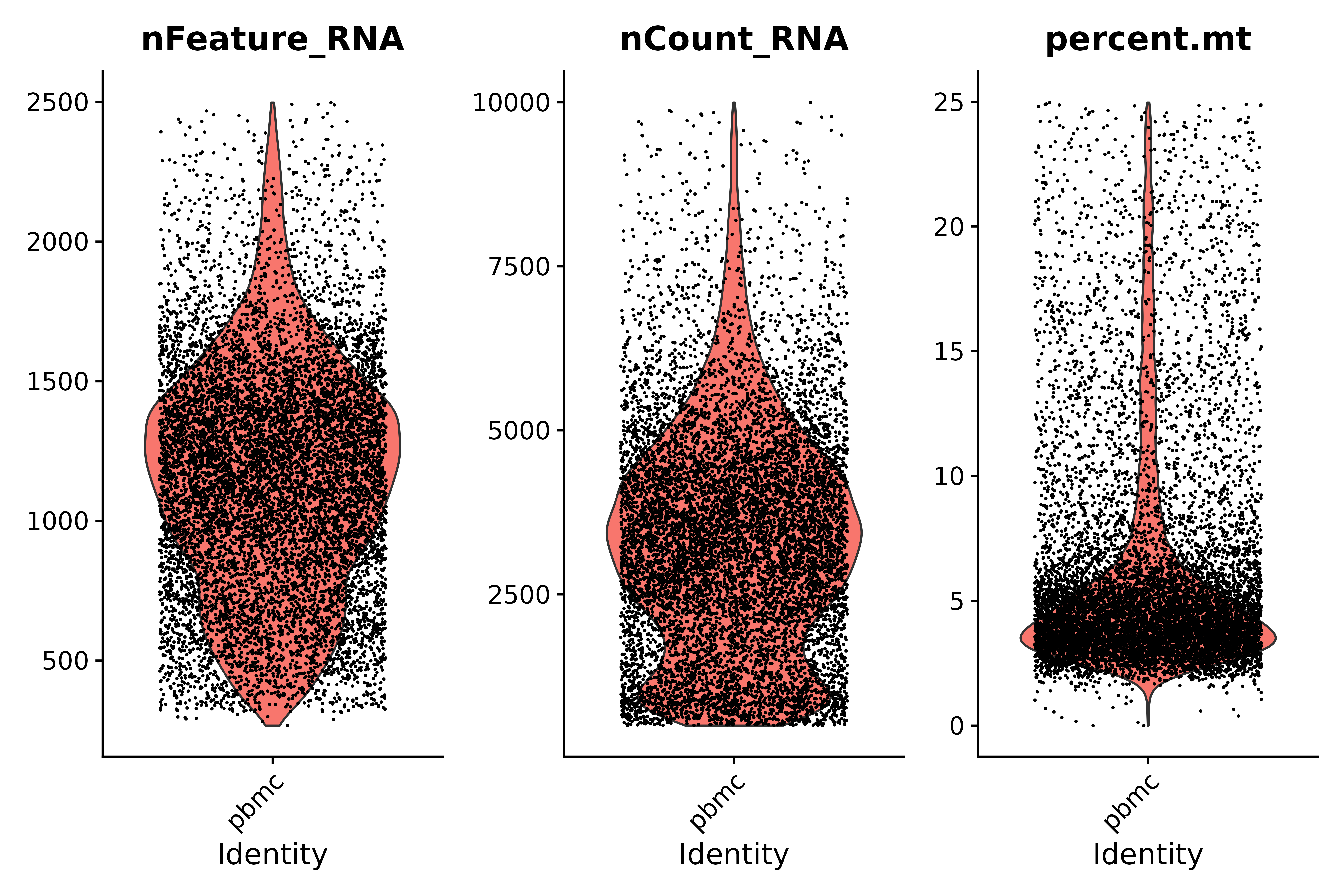
4. Normaliser les données et identifier les caractéristiques très variables (sélection des caractéristiques)
Dans l’analyse des données du transcriptome unicellulaire, nous effectuons d’abord une normalisation des données pour garantir que les niveaux d’expression génétique entre différentes cellules peuvent être comparés efficacement.
1. Normalisation des données
- Normalisation des données : nous divisons d’abord le niveau d’expression (nombre) d’un gène spécifique dans chaque cellule par le niveau d’expression total (nombre total) de la cellule. Cette étape vise à éliminer la différence de profondeur de séquençage entre les cellules afin que les niveaux d’expression génétique de différentes cellules puissent être comparés sur la même base.
- Ajustement de l'échelle : la valeur d'expression normalisée est ensuite multipliée par un facteur d'échelle, ici 10 000, afin d'ajuster la valeur d'expression du gène à une grandeur plus appropriée pour l'analyse ultérieure.
- Transformation logarithmique : Enfin, nous avons effectué une transformation logarithmique sur les valeurs d'expression ajustées, ce qui a contribué à stabiliser davantage la variance et à rendre les différences entre les gènes fortement et faiblement exprimés plus évidentes, jetant les bases des étapes d'analyse ultérieures.
- Normalisation : Grâce aux étapes ci-dessus, nous avons normalisé les valeurs d’expression génétique au niveau de 10 000 UMI, ce qui permet d’éliminer l’impact des différences de profondeur de séquençage entre les cellules.
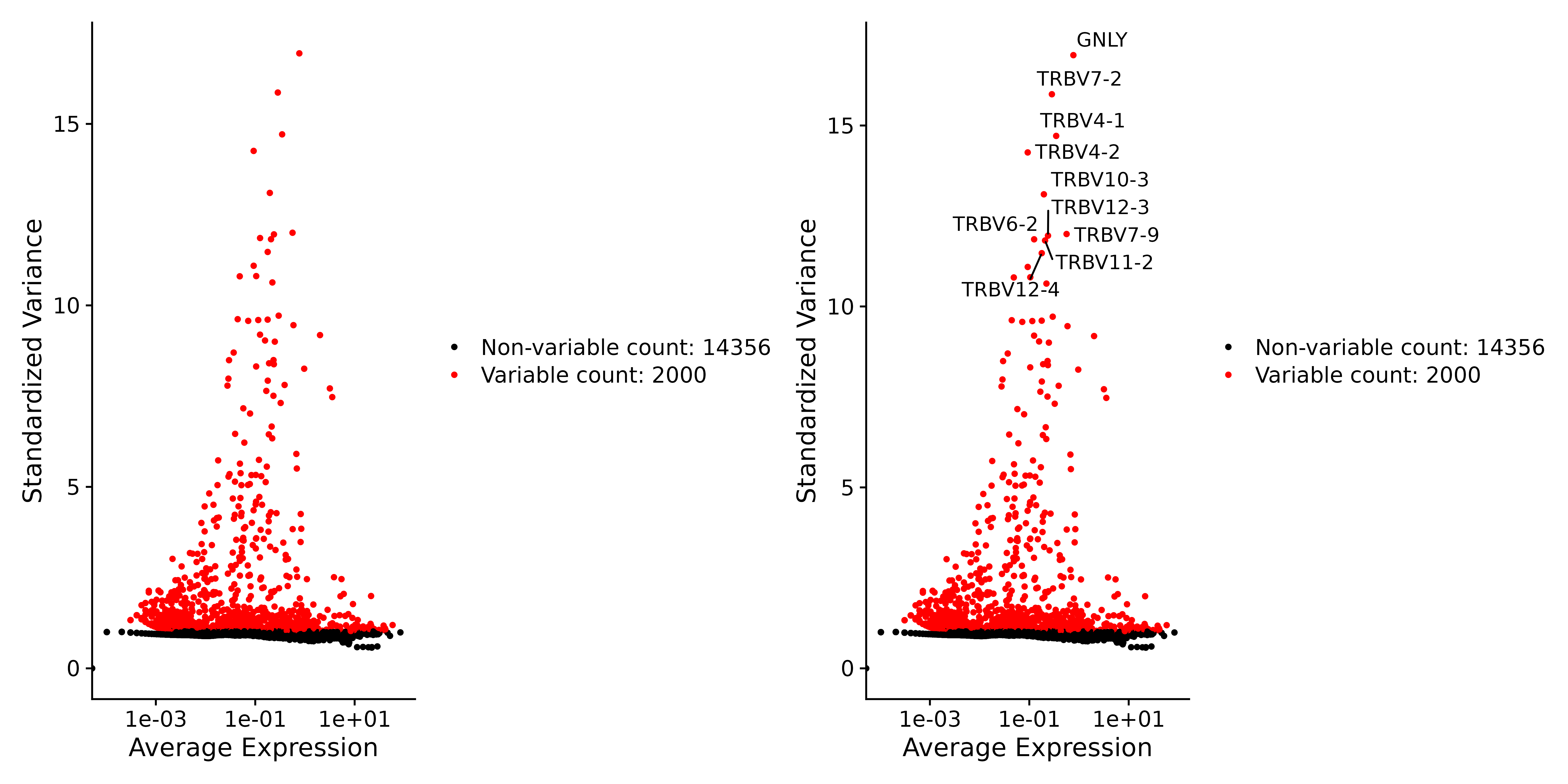
2. Ensuite, nous recherchons des caractéristiques de mutation :
Après normalisation et standardisation, nous avons identifié les gènes hypervariables en analysant la variabilité de l’expression des gènes. Ces gènes ont des niveaux d’expression significativement différents dans différentes cellules et peuvent être fortement exprimés dans certaines cellules mais exprimés à des niveaux inférieurs dans d’autres.
# IV. Normalize Data----------------------------------------------------------------
seurat.subset <- NormalizeData(seurat.subset, normalization.method = "LogNormalize", scale.factor = 10000)
seurat.subset <- FindVariableFeatures(seurat.subset, selection.method = "vst", nfeatures = 2000)
# Identify the 10 most highly variable genes
top10 <- head(VariableFeatures(seurat.subset), 10)
# plot variable features with and without labels
plot3 <- VariableFeaturePlot(seurat.subset)
plot4 <- LabelPoints(plot = plot3, points = top10, repel = TRUE)
ggsave("VariableFeaturePlot_4.png", plot3 + plot4, width = 12, height = 6, dpi=600)
5. Mettre les données à l'échelle et exécuter l'ACP
Dans l'analyse des données du transcriptome unicellulaire, la mise à l'échelle des données et l'ACP peuvent réduire efficacement la dimension des données, extraire les principales sources de variation intercellulaire et fournir des informations importantes pour comprendre l'hétérogénéité et la fonction des cellules.
1. Mise à l'échelle des données : la mise à l'échelle des données est une transformation linéaire qui vise à convertir les données d'expression génétique en une distribution normale standard, c'est-à-dire que la moyenne des données est de 0 et l'écart type de 1. Cette transformation permet de réduire les différences entre les niveaux d'expression des différents gènes et rend les données plus normalisées. Grâce à la mise à l’échelle, l’impact des valeurs extrêmes ou des valeurs aberrantes dans les données peut être réduit, ce qui rend la distribution des données plus uniforme et fournit des données plus stables pour les étapes d’analyse ultérieures. Cette étape est également une préparation à l’ACP (Analyse en Composantes Principales), car l’ACP est plus performante sur des données standardisées.
2. Exécutez PCA :
- L'ACP est une méthode statistique qui transforme les données dans un nouveau système de coordonnées en transformant les axes orthogonaux de sorte que la première plus grande variance de toute projection des données soit sur la première coordonnée (appelée première composante principale), la deuxième plus grande variance soit sur la deuxième coordonnée, et ainsi de suite.
- Dans l'analyse du transcriptome unicellulaire, l'ACP est utilisée pour séparer les cellules individuelles en fonction des différences d'expression génétique. Chaque cellule peut être considérée comme un point dans un espace de grande dimension, et l'ACP représente à nouveau ces points dans un espace de faible dimension tout en conservant autant d'informations de variation que possible.
- Chaque composante principale (PCA) représente essentiellement une « méta-caractéristique » de la cellule, c'est-à-dire une direction majeure de variation de la cellule. Dans les résultats de l’ACP, plus la composante principale est avancée, plus son poids dans la variation des données est important, ce qui signifie qu’elle contient plus d’informations.
- Grâce à l'ACP, les informations d'expression de haute dimension des cellules peuvent être compressées en plusieurs composants principaux clés, qui peuvent être utilisés pour une analyse de clustering ultérieure, une visualisation ou d'autres analyses en aval.
all.genes <- rownames(seurat.subset)
seurat.subset <- ScaleData(seurat.subset, features = all.genes)
seurat.subset <- RunPCA(seurat.subset, features = VariableFeatures(object = seurat))##### Examine and visualize PCA results a few different ways-------------
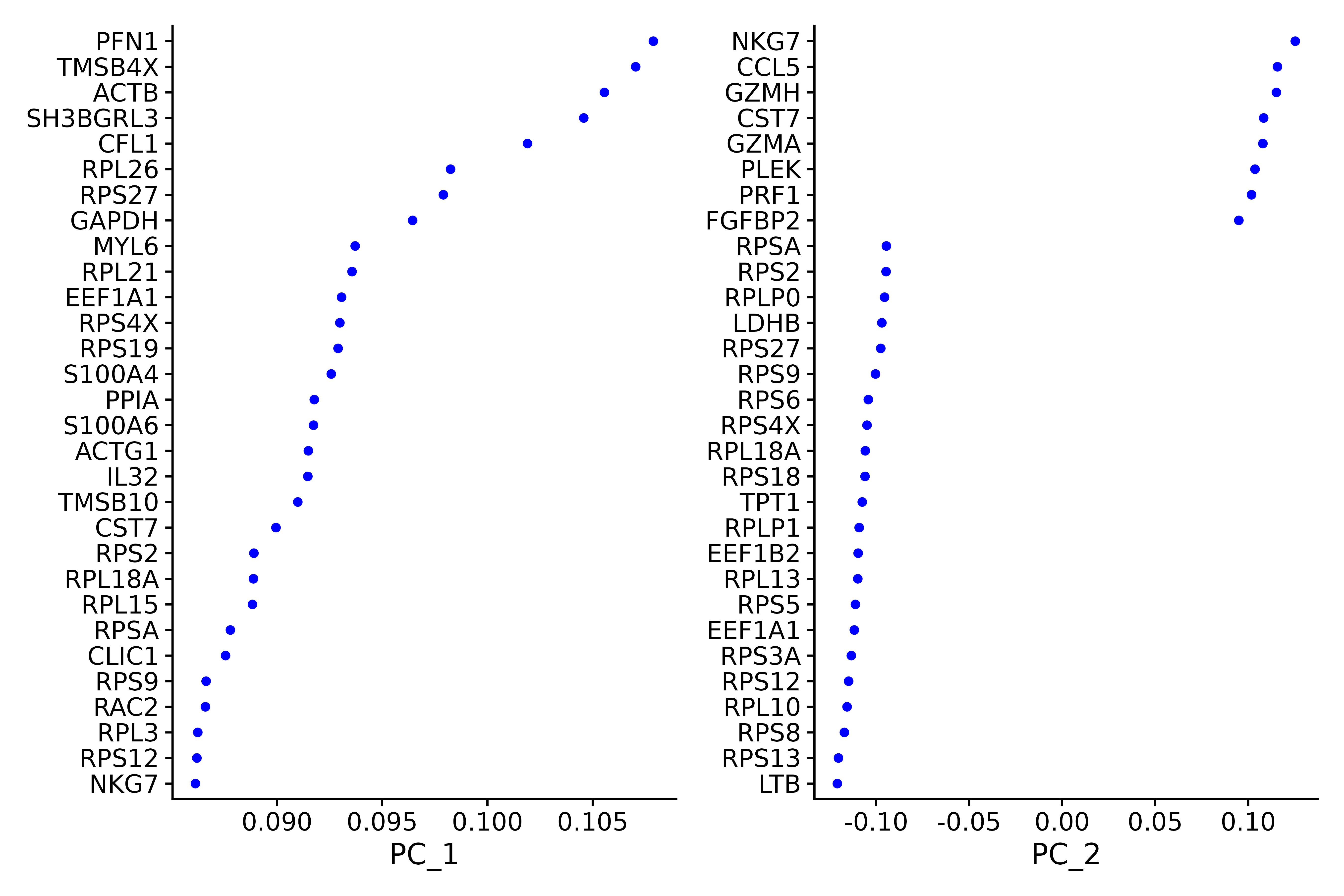
print(seurat.subset[["pca"]], dims = 1:5, nfeatures = 5)
p1 <- VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
p1
ggsave("VizDimLoadings_5.png", p1, width = 9, height = 6, dpi=600)
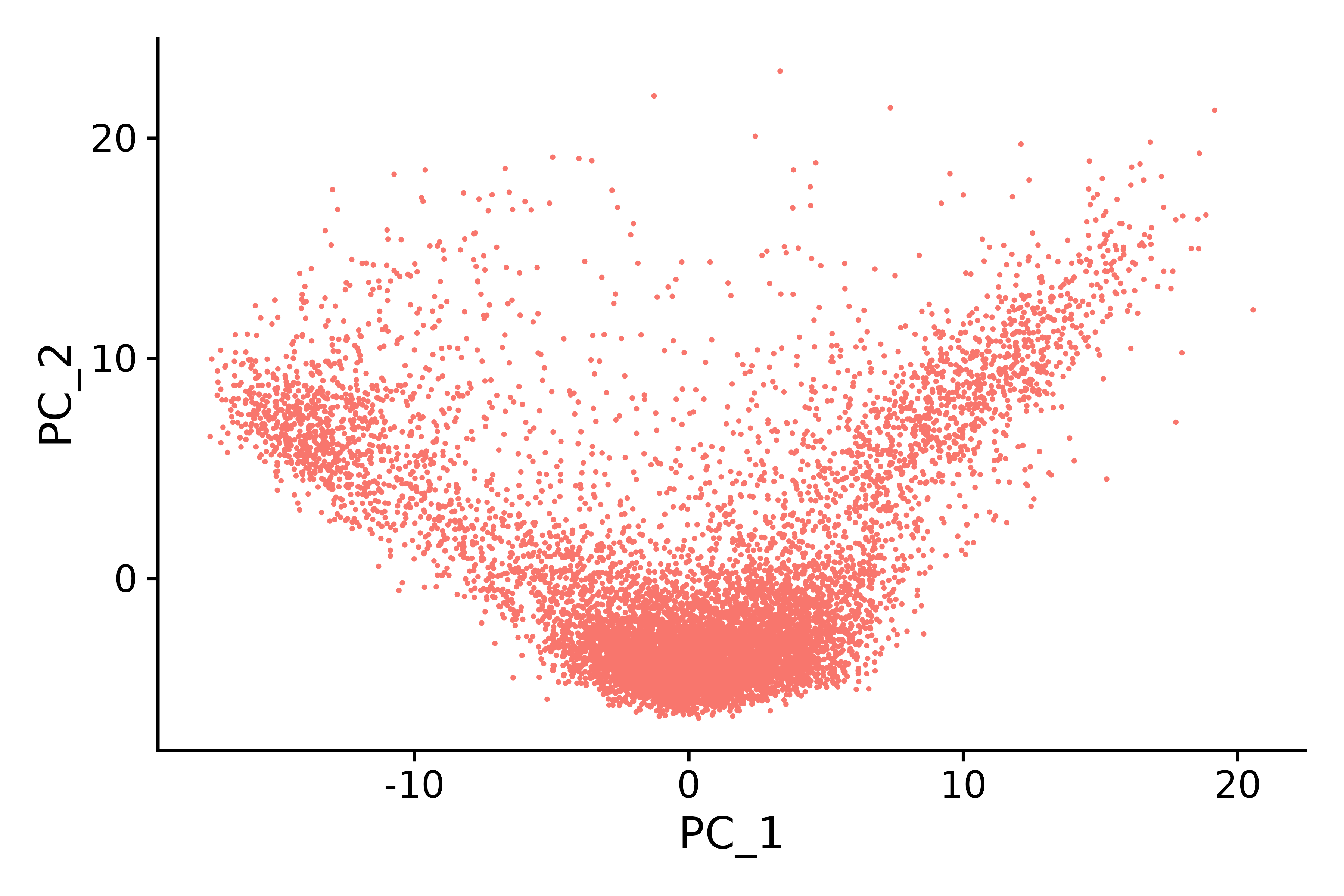
p2 <- DimPlot(seurat.subset, reduction = "pca") + NoLegend()
p2
ggsave("pca_6.png", p2, width = 6, height = 4, dpi=600)
p3 <- DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
p3
ggsave("pca_1dims.png", p3, width = 6, height = 4, dpi=600)
p4 <- DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
p4
ggsave("pca_15dims.png", p4, width = 10, height = 10, dpi=600)
p5 <- ElbowPlot(seurat.subset)
p5
ggsave("ElbowPlot.png", p5, width = 10, height = 10, dpi=600)
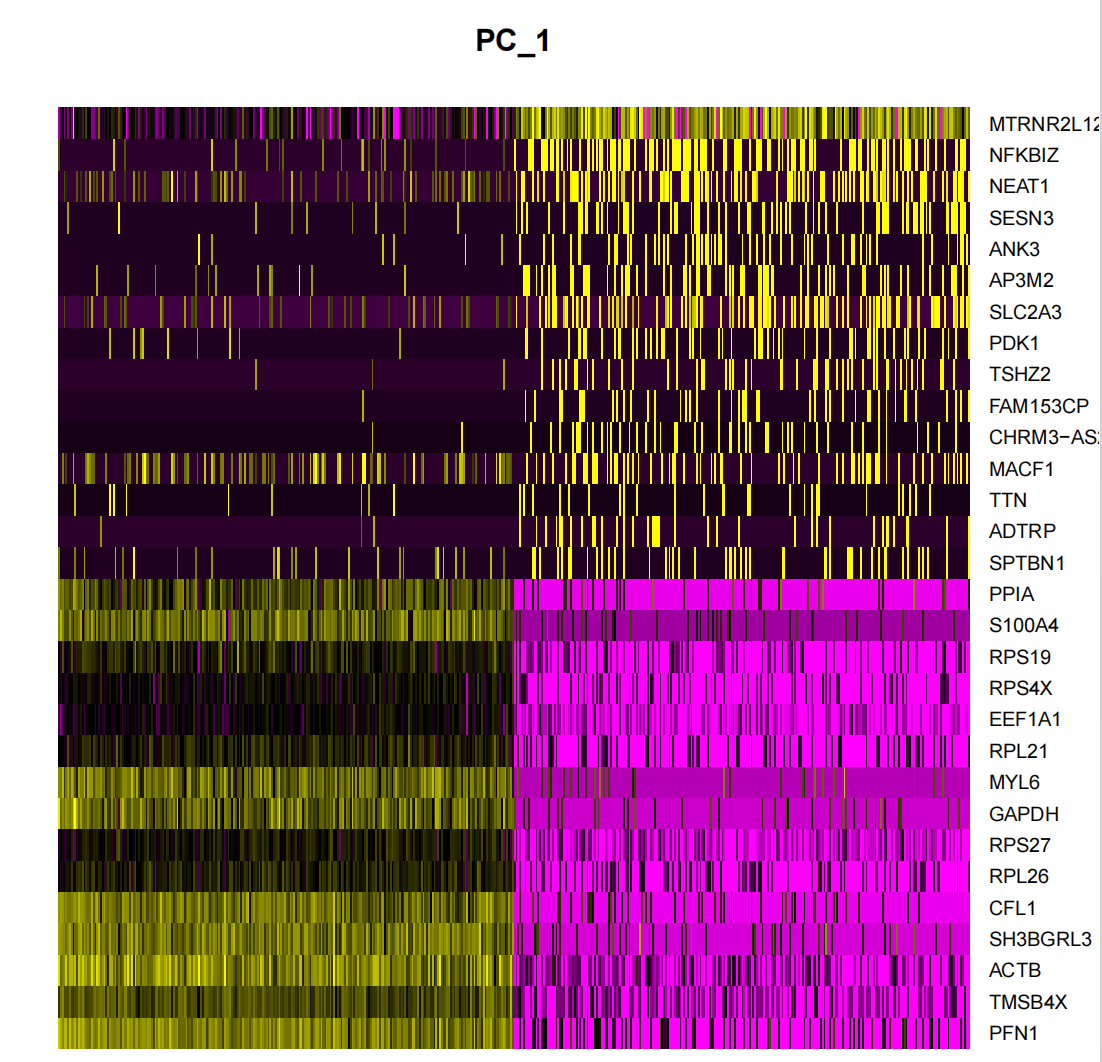
DimHeatmap() est un puissant outil d'analyse de données qui permet aux chercheurs d'identifier et d'explorer facilement les principales sources d'hétérogénéité dans un ensemble de données. Après avoir effectué une analyse en composantes principales (ACP), DimHeatmap() est particulièrement utile pour déterminer quels composants principaux (PC) doivent être inclus pour éclairer l'analyse approfondie ultérieure. Ce processus implique le classement des cellules et des caractéristiques en fonction des scores PCA, garantissant ainsi l’exactitude et la pertinence de l’analyse.
De plus, DimHeatmap() permet à l'utilisateur de définir un nombre spécifique de cellules pour tracer spécifiquement les cellules « extrêmes » qui présentent des caractéristiques extrêmes aux deux extrémités du spectre de données. Cette approche améliore considérablement l’efficacité du traçage lorsque vous travaillez avec de grands ensembles de données, car elle se concentre sur les parties les plus saillantes des données, accélérant ainsi l’ensemble du processus de visualisation.

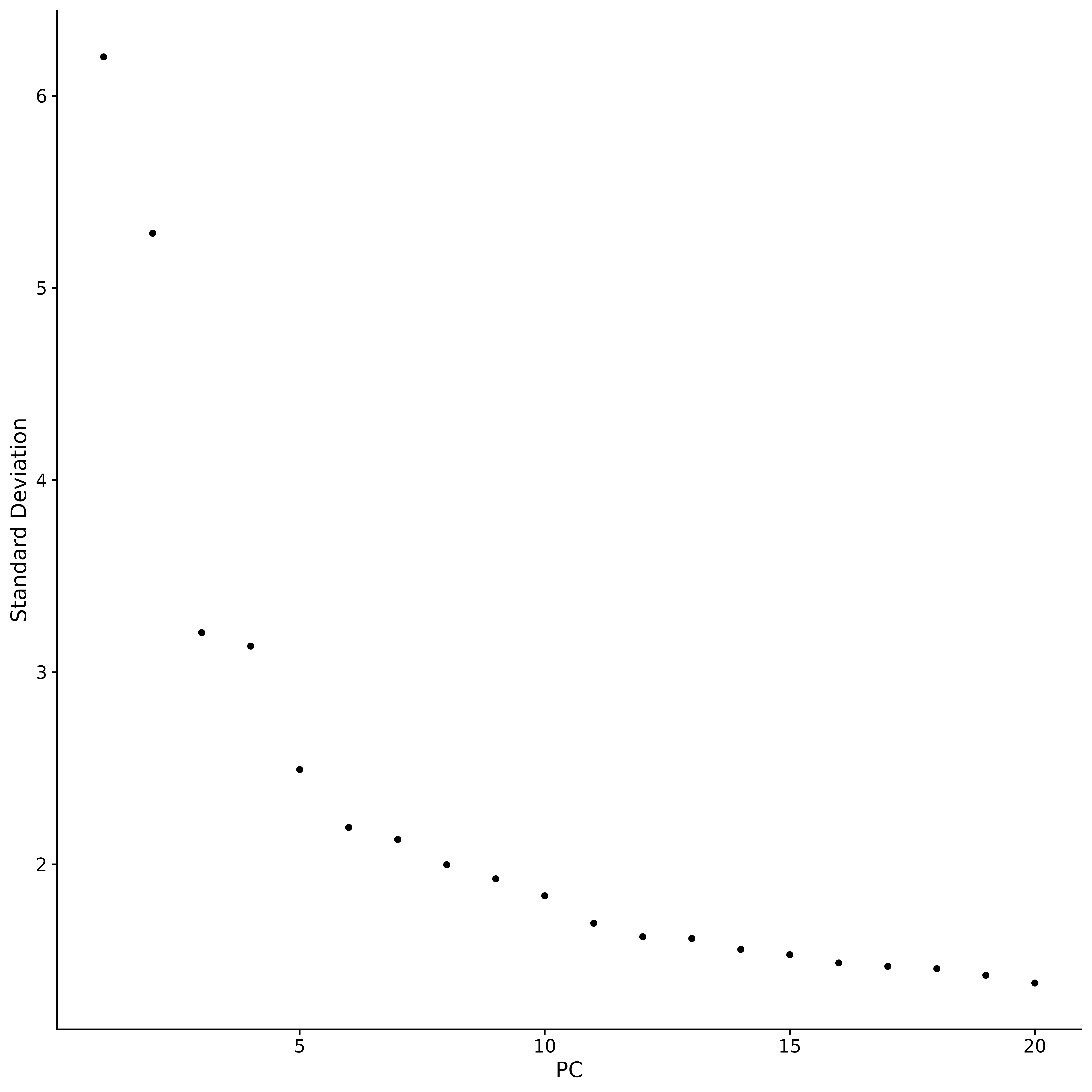
Il est courant dans l’analyse en composantes principales (ACP) d’utiliser le « diagramme en coude » pour déterminer le nombre de composantes principales à conserver pour l’analyse ultérieure. Le graphique montre que la majeure partie du vrai signal est capturée dans les 17 premiers composants principaux. Dans le graphique du coude, nous recherchons un point de retournement, le « coude ». Avant ce point, chaque composante principale supplémentaire améliore considérablement le pouvoir explicatif des données, mais après ce point, le gain résultant de l’ajout de composantes principales commence à diminuer.Cela indique qu’un état d’équilibre a été atteint.. Sur la base des résultats de l’analyse du graphique du coude, nous avons sélectionné les 17 premiers composants principaux pour une analyse ultérieure. Parce que l’ajout de composants principaux supplémentaires n’améliorera pas significativement la discrimination des données, mais entraînera une augmentation inutile de la quantité de calcul. Si nous continuons à augmenter le nombre de composantes principales, la discrimination des données peut être légèrement améliorée, mais cette amélioration n’est pas significative et entraînera une augmentation significative des coûts de calcul. Par conséquent, choisir 17 composantes principales est un choix raisonnable qui équilibre la discrimination et l’efficacité de calcul.
pdf("dimreduction_plots.pdf")#保存为 pdf 文件
VizDimLoadings(seurat.subset, dims = 1:2, reduction = "pca")
DimPlot(seurat.subset, reduction = "pca")
DimHeatmap(seurat.subset, dims = 1, cells = 500, balanced = TRUE)
DimHeatmap(seurat.subset, dims = 1:15, cells = 500, balanced = TRUE)
ElbowPlot(seurat.subset)
dev.off()seurat.subset.dimReduction <- seurat.subset
seurat.subset.dimReduction <- FindNeighbors(seurat.subset.dimReduction, reduction="pca", dims = 1:17)
saveRDS(seurat.subset.dimReduction, file = "~/home/output/data/seurat_pre_FindClusters.rds")
# seurat.subset.dimReduction <- readRDS("~/home/output/data/seurat_pre_FindClusters.rds")6. Regroupement de cellules
### 3. Find clusters,聚类
```{r find_clusters}
seurat.final <- FindClusters(object = seurat.subset.dimReduction, resolution = 0.1)
head(Idents(seurat.final), 5)
seurat.final <- RunUMAP(seurat.final, reduction="pca", dims=1:17)
#Visualize with UMAP
p1<- DimPlot(seurat.final, reduction = "umap")
p1
ggsave("find_clusters.png", p1, width = 6, height =5 , dpi=600)
saveRDS(seurat.final, file = "~/home/output/data/seurat_final.rds")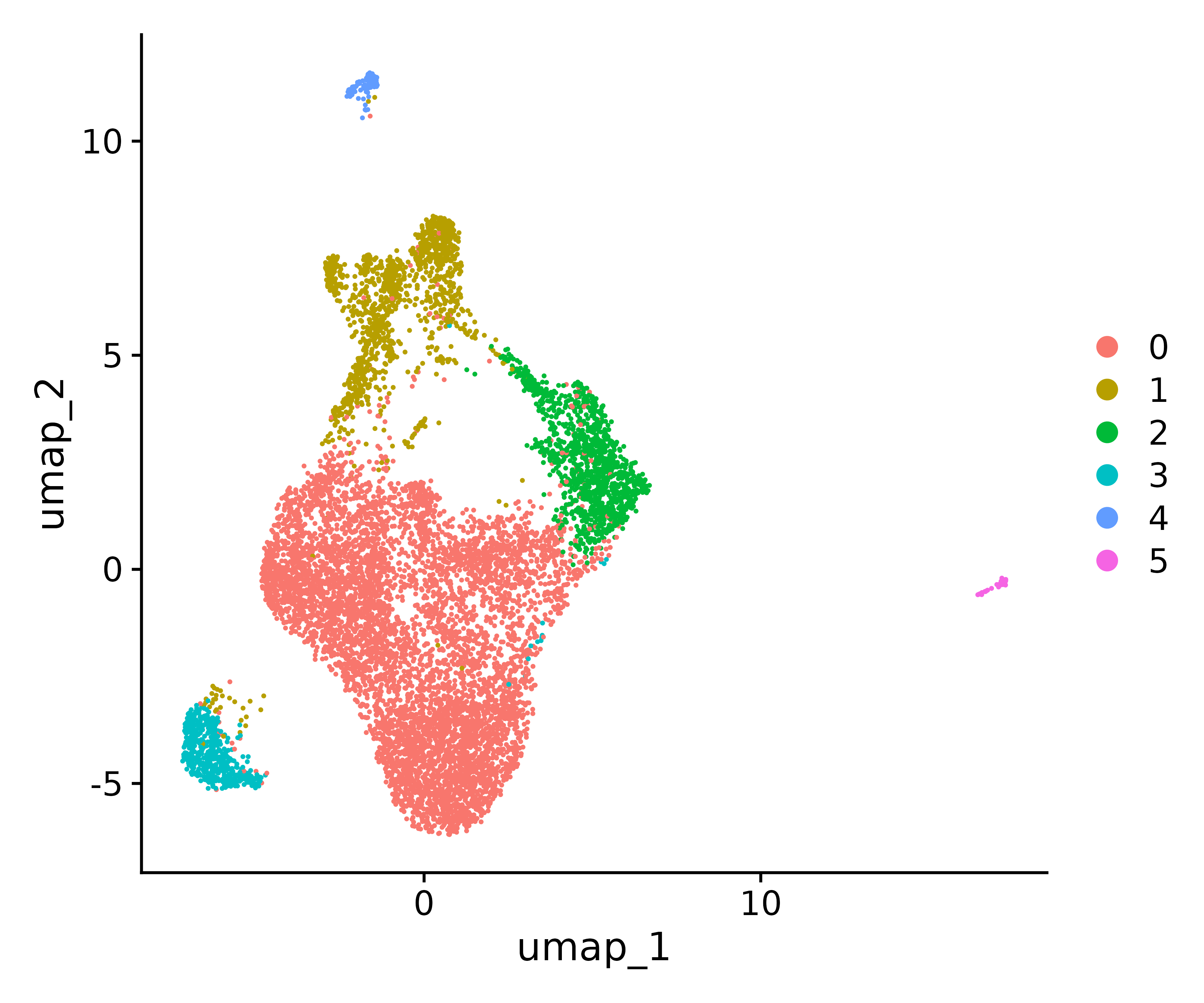
Visualisation
Il existe trois graphiques courants pour visualiser l’expression des gènes dans différents groupes :
1. Carte thermique 2. Carte à bulles 3. Graphique des caractéristiques
table(Idents(seurat.final))
# find markers between cluster1 and cluster 2
##用来找 cluster 之间的寻找差异表达特征
cluster2.markers <- FindMarkers(seurat.final, ident.1 = 1,ident.2 = 2)
head(cluster2.markers, n = 5)
# find markers for every cluster compared to all remaining cells, report only the positive ones
#找每个 cluster 的 marker 基因
pbmc.markers <- FindAllMarkers(seurat.final, only.pos = TRUE)
ClusterMarker_noRibo <- pbmc.markers[!grepl("^RP[SL]", pbmc.markers$gene, ignore.case = F),]
ClusterMarker_noRibo_noMito <- ClusterMarker_noRibo[!grepl("^MT-", ClusterMarker_noRibo$gene, ignore.case = F),]
###去除核糖体基因和线粒体基因的影响
ClusterMarker_noRibo_noMito %>%
group_by(cluster) %>%
dplyr::filter(avg_log2FC > 1) %>%
slice_head(n = 5) %>%
ungroup() -> top5
###展示每个 cluster 的 top5 的基因
heatmap <- DoHeatmap(seurat.final, features = top5$gene) + NoLegend()
ggsave("heatmap.png", heatmap, width = 10, height =5 , dpi=600) Cette figure est une carte thermique montrant les gènes marqueurs
Cette figure est une carte thermique montrant les gènes marqueurs
g =unique(top5$gene)
##定义 top5 的 gene 列,作为基因集 g,也可以自定义
p <- DotPlot(seurat.final,features=g)+
coord_flip() + # 翻转坐标轴
theme(
panel.grid = element_blank(), # 移除网格线
axis.text.x = element_text(angle = 0, hjust = 0.5, vjust = 0.5) # 旋转 x 轴标签
)
ggsave("allmarker_dotpot.png", p, width = 8, height =7 , dpi=600)
#气泡图展示 marker 基因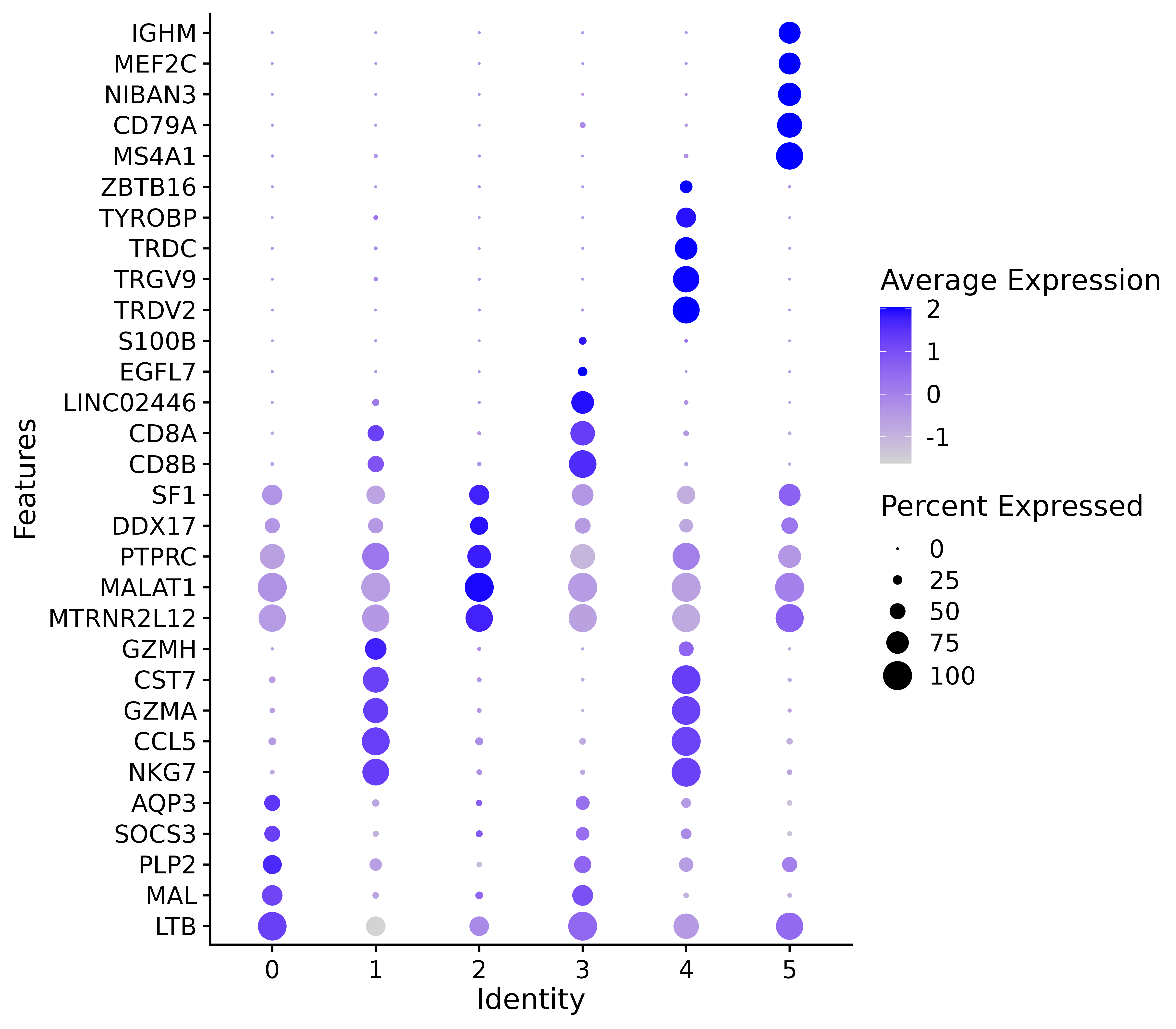
Cette figure est un graphique à bulles montrant les gènes marqueurs
# 加载 dplyr 包
library(dplyr)
# 提取 cluster 和 gene 列
cluster_gene_mapping <- top5 %>%
select(cluster, gene)
# 查看结果
head(cluster_gene_mapping,30)
write.csv(cluster_gene_mapping,"cluster_allmarkers_TOP5.csv", row.names = p <- FeaturePlot(seurat.final, features = g)
ggsave("allmarker_featurepot.png", p, width = 15, height =15 , dpi=600)
###查看自定义感兴趣的基因
p2 <- FeaturePlot(seurat.final, features = c("MS4A1", "GNLY", "CD3E", "CD14", "FCER1A", "FCGR3A", "LYZ", "PPBP",
"CD8A"))
ggsave("allmarker_featurepot2.png", p2, width = 15, height =15 , dpi=600)


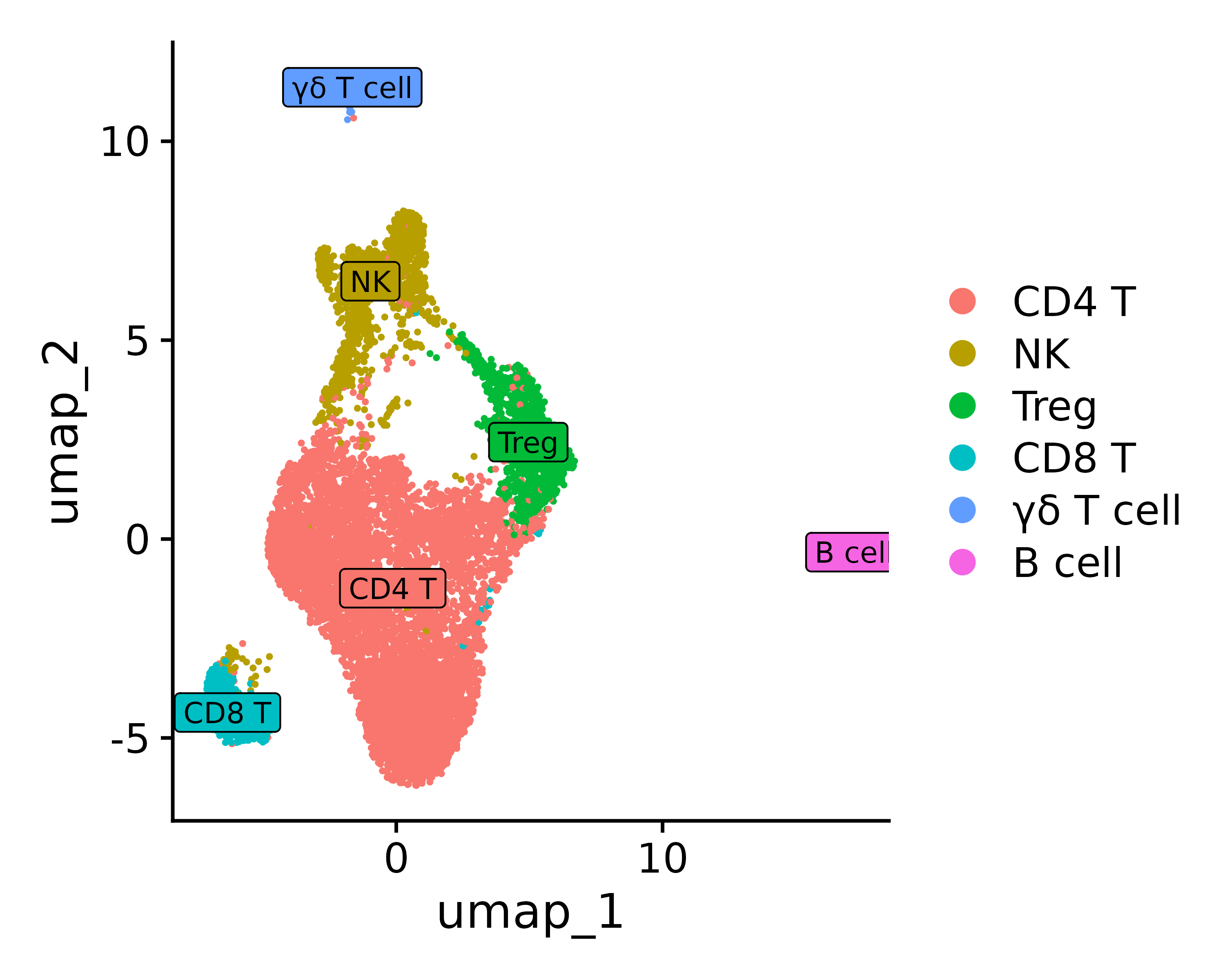
7. Analyse des données
Cette étape décrit comment utiliser des gènes marqueurs spécifiques pour déterminer et définir les types de cellules dans l’analyse des données du transcriptome unicellulaire.
En supposant que vous ayez déterminé le type de cellule en fonction des gènes marqueurs supérieurs, etc., vous pouvez définir le type d'identité
0 1 2 3 4 5 « CD4 T », « NK », « Treg », « CD8 T », « lymphocyte T γδ », « lymphocyte B »
(Les commentaires suivants sont fournis à titre indicatif uniquement et ne sont pas des commentaires réels.)
new.cluster.ids <- c("CD4 T", "NK", "Treg", "CD8 T", "γδ T cell",
"B cell")
names(new.cluster.ids) <- levels(seurat.final)
seurat.final <- RenameIdents(seurat.final, new.cluster.ids)
p <- DimPlot(seurat.final, reduction = "umap", label = TRUE, pt.size = 0.5) + NoLegend()
p3 = DimPlot(seurat.final, reduction = "umap", label = TRUE, label.size = 3, pt.size = .3, label.box = T)
ggsave("annotation_umap.png", p, width = 5, height =4 , dpi=600)
ggsave("annotation_umap2.png", p3, width = 5, height =4 , dpi=600)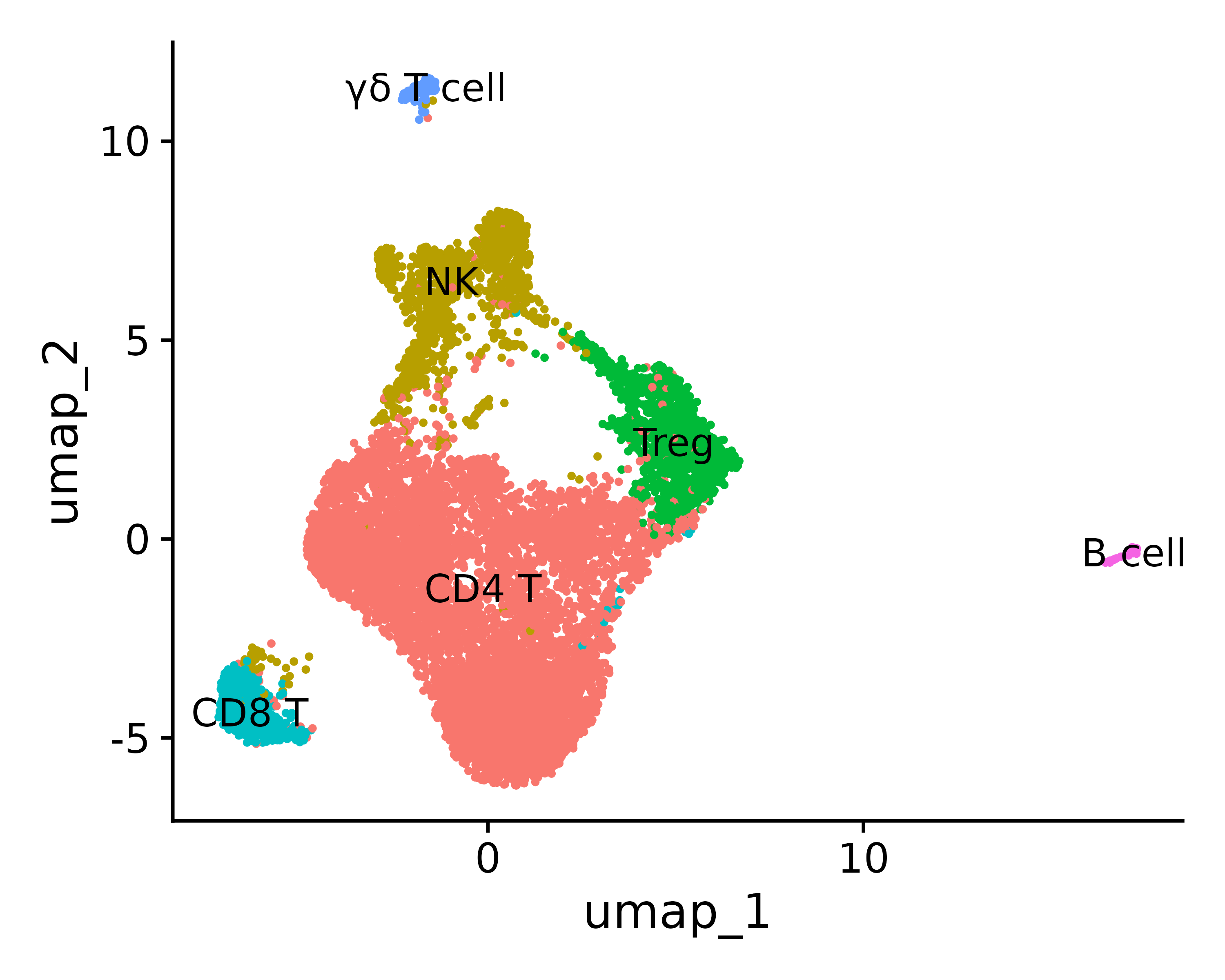
Référence
Créer de l'IA avec l'IA
De l'idée au lancement — accélérez votre développement IA avec le co-codage IA gratuit, un environnement prêt à l'emploi et le meilleur prix pour les GPU.