Command Palette
Search for a command to run...
Tutoriel De Démarrage GROMACS : Lysozyme Dans l'eau
Simulation de dynamique moléculaire en utilisant le « lysozyme dans l'eau » comme exemple
Introduction au tutoriel
Ce tutoriel est un tutoriel d'introduction à la simulation de dynamique moléculaire à l'aide du logiciel GROMACS. Lysozyme dans l'eau À titre d’exemple, apprenez à préparer et à exécuter une simulation de dynamique moléculaire d’une protéine typique dans l’eau.
La version GPU de GROMACS utilisant la carte graphique NVIDIA RTX 4090 sur la plate-forme OpenBayes a considérablement amélioré son efficacité de calcul après le calcul parallèle GPU, avec une vitesse allant jusqu'à 255 ns/jour ! Voici les performances de vitesse :
Core t (s) Wall t (s) (%)
Time: 3972.923 198.659 1999.9
(ns/day) (hour/ns)
Performance: 255.471 0.094
Introduction à GROMACS
GROMACS (GROningen MAchine for Chemical Simulations) est un progiciel hautes performances pour les simulations de dynamique moléculaire. Il est principalement utilisé pour modéliser et simuler le comportement de mouvement des molécules biologiques (telles que les protéines, les lipides et les acides nucléiques) dans différentes conditions. Il a été développé à l'origine à l'Université de Groningue aux Pays-Bas et est devenu l'un des outils open source les plus couramment utilisés dans le domaine de la dynamique moléculaire.
Principales caractéristiques de GROMACS
1. haute performance:
• GROMACS est hautement optimisé pour le calcul parallèle et peut fonctionner efficacement sur les systèmes CPU et GPU multicœurs modernes.
• Prend en charge OpenMP et MPI pour le calcul multithread et distribué.
2. Large gamme d'applications:
• Peut être utilisé pour simuler le comportement de petites molécules jusqu'à de grands complexes protéiques.
• Soutient la recherche sur les biomolécules, les polymères, les composés inorganiques et d’autres systèmes chimiques.
3. flexibilité:
• Fournit un ensemble riche d'outils pour le prétraitement (par exemple, la génération de topologie, la solvatation) et le post-traitement (par exemple, l'analyse de trajectoire, le calcul d'énergie).
• Prise en charge de divers champs de force, tels que AMBER, CHARMM et GROMOS.
4. Facilité d'utilisation:
• GROMACS comprend une interface de ligne de commande conviviale.
• Fournit une documentation détaillée et des tutoriels adaptés aux utilisateurs débutants et avancés.
5. Open source et évolutif:
• La licence open source permet aux utilisateurs de modifier et d’étendre GROMACS pour répondre à des besoins spécifiques.
• Dispose d’une communauté d’utilisateurs active et d’une équipe de développement.
Processus d'utilisation générale de GROMACS
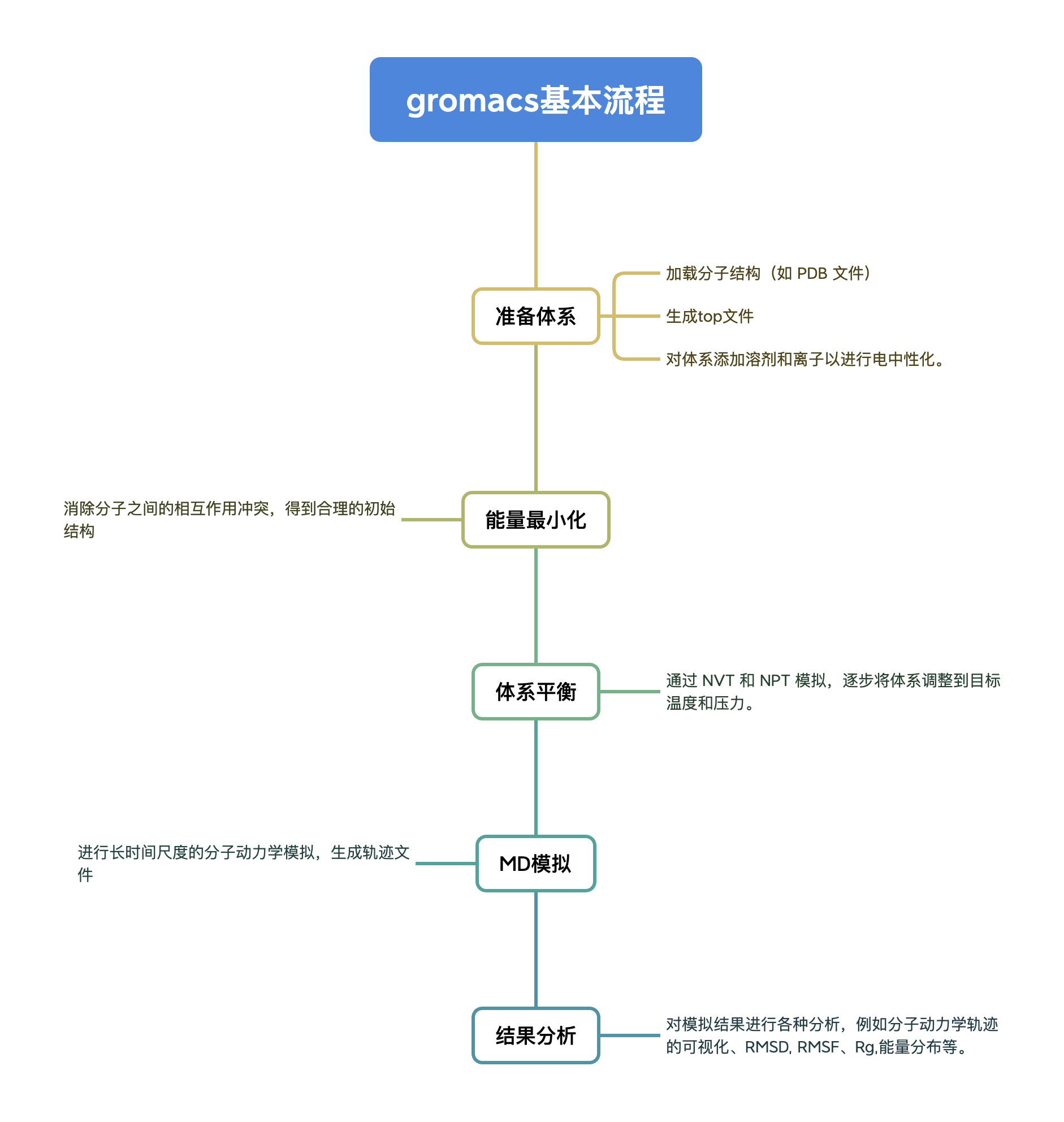
Étapes de course
Nous devons d'abord configurer le fichier de protéines pdb et le fichier de calcul md, puis après divers prétraitements, nous effectuons enfin une simulation de 10 ns et analysons les résultats. Voici une explication étape par étape.
1. Démarrez GROMACS
首先登录平台:https://openbayes.com/
选择「高性能计算」> 创建新容器> 选择算力 RTX 4090> 选择 gromacs GPU
打开工作空间
打开终端
或者采用 SSH 控制服务器:
在 X-shell 或者 mac unix 终端,输入:ssh -p 32699 [email protected],再输入密码即可(如下所示)↓
liangzhongzhongzhong@lzr ~ % ssh -p 32699 [email protected]
The authenticity of host '[ssh.openbayes.com]:32699 ([101.237.34.75]:32699)' can't be established.
ED25519 key fingerprint is SHA256:uwPyhP/EYoW49Ez4rvAuaf19czwis2rdS4pImsR0NH8.
This key is not known by any other names.
Are you sure you want to continue connecting (yes/no/[fingerprint])? yes
Warning: Permanently added '[ssh.openbayes.com]:32699' (ED25519) to the list of known hosts.
[email protected]'s password:
OpenBayes
目录说明
- /openbayes/home 工作空间内的数据保存地址,容器停止后,该目录中的内容不会被删除
- /openbayes/input/input0 - /openbayes/input/input4 为数据目录,不会占用工作空间的存储容量,最多支持同时绑定 5 个
⚠️ 其他目录下的内容在容器关闭后会被自动删除!更多信息请访问 https://openbayes.com/docs/concepts
⚠️ 禁止挖矿,一经发现将立即封号恕不退款
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/home# ls
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes# l(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes# lhome input 请将文件存在 home 目录下, 当前文件夹下的文件不会被保存.txt
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes# ls(base) root@liangzhong-4ay9ej85pxvd-main:/openbaye
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/input# cd input0
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/input/input0# ls
'#nvt.log.1#' '#topol.top.2#' 1AKI_processed.gro 1aki.pdb em.log ions.mdp mdout.mdp nvt.cpt nvt.log nvt.trr topol.top
'#nvt.log.2#' 1AKI_clean.pdb 1AKI_solv.gro em.edr em.tpr ions.tpr minim.mdp nvt.edr nvt.mdp posre.itp
'#topol.top.1#' 1AKI_newbox.gro 1AKI_solv_ions.gro em.gro em.trr md.mdp npt.mdp nvt.gro nvt.tpr potential.xvg
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/input/input0#

调用 GROMACS,设置临时环境变量
export PATH=/data/app/gromacs/bin:$PATH
(base) root@liangzhong-4ay9ej85pxvd-main:/openbayes/home# gmx_mpi -h
:-) GROMACS - gmx_mpi, 2023 (-:
Executable: /data/app/gromacs/bin/gmx_mpi
Data prefix: /data/app/gromacs
Working dir: /output
Command line:
gmx_mpi -h
SYNOPSIS
gmx [-[no]h] [-[no]quiet] [-[no]version] [-[no]copyright] [-nice <int>]
[-[no]backup]
OPTIONS
Other options:
-[no]h (no)
Print help and quit
-[no]quiet (no)
Do not print common startup info or quotes
-[no]version (no)
Print extended version information and quit
-[no]copyright (no)
Print copyright information on startup
-nice <int> (19)
Set the nicelevel (default depends on command)
-[no]backup (yes)
Write backups if output files exist
Additional help is available on the following topics:
commands List of available commands
selections Selection syntax and usage
To access the help, use 'gmx help <topic>'.
For help on a command, use 'gmx help <command>'.
GROMACS reminds you: "All You Need is Greed" (Aztec Camera)
2. Préparation des documents
Avant d'exécuter le tutoriel, nous devons préparer les 6 fichiers suivants : 1aki.pdb, ions.mdp, md.mdp, minim.mdp, npt.mdp, nvt.mdp
Ces fichiers peuvent être téléchargés directement ou créés comme suit.
Par exemple, le fichier de protéines 1AKI.pdb peut être obtenu à partir de RCSB Obtenez 1AKI.pdb à partir du site Web.Dans le didacticiel, d'autres fichiers ont été créés en copiant le code à l'aide de l'éditeur de texte vim.
Introduction à la base de données RCSB :
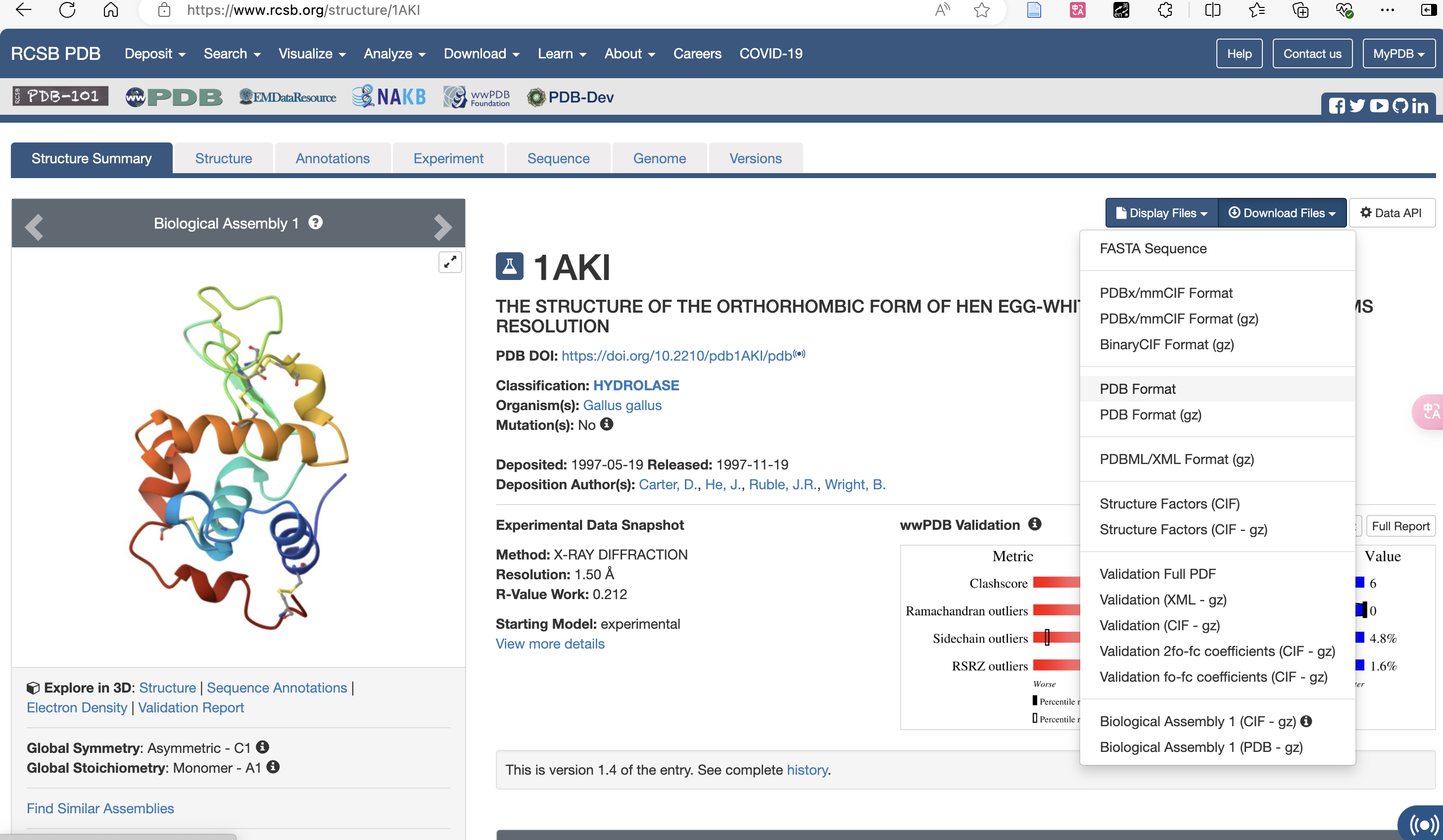
Télécharger le format pdb :
Téléchargez dans votre répertoire de travail
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# ls
1aki.pdb
#查看上传成功
vim ions.mdp
#准备 mdp 文件,输入命令后复制一下内容,按 i 进入插入模式开始编辑。
#编辑完成后,按 Esc 键退出插入模式,输入 :wq 保存并退出。
; ions.mdp - used as input into grompp to generate ions.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Minimization step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; Method to determine neighbor list (simple, grid)
coulombtype = cutoff ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; Short-range electrostatic cut-off
rvdw = 1.0 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions in all 3 dimensions
vim minim.mdp
; minim.mdp - used as input into grompp to generate em.tpr
; Parameters describing what to do, when to stop and what to save
integrator = steep ; Algorithm (steep = steepest descent minimization)
emtol = 1000.0 ; Stop minimization when the maximum force < 1000.0 kJ/mol/nm
emstep = 0.01 ; Minimization step size
nsteps = 50000 ; Maximum number of (minimization) steps to perform
; Parameters describing how to find the neighbors of each atom and how to calculate the interactions
nstlist = 1 ; Frequency to update the neighbor list and long range forces
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; Method to determine neighbor list (simple, grid)
coulombtype = PME ; Treatment of long range electrostatic interactions
rcoulomb = 1.0 ; Short-range electrostatic cut-off
rvdw = 1.0 ; Short-range Van der Waals cut-off
pbc = xyz ; Periodic Boundary Conditions in all 3 dimensions
vim nvt.mdp
title = OPLS Lysozyme NVT equilibration
define = -DPOSRES ; position restrain the protein
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 500 ; save coordinates every 1.0 ps
nstvout = 500 ; save velocities every 1.0 ps
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
; Bond parameters
continuation = no ; first dynamics run
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Nonbonded settings
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
DispCorr = EnerPres ; account for cut-off vdW scheme
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling is off
pcoupl = no ; no pressure coupling in NVT
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Velocity generation
gen_vel = yes ; assign velocities from Maxwell distribution
gen_temp = 300 ; temperature for Maxwell distribution
gen_seed = -1 ; generate a random seed
vim npt.mdp
title = OPLS Lysozyme NPT equilibration
define = -DPOSRES ; position restrain the protein
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 50000 ; 2 * 50000 = 100 ps
dt = 0.002 ; 2 fs
; Output control
nstxout = 500 ; save coordinates every 1.0 ps
nstvout = 500 ; save velocities every 1.0 ps
nstenergy = 500 ; save energies every 1.0 ps
nstlog = 500 ; update log file every 1.0 ps
; Bond parameters
continuation = yes ; Restarting after NVT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Nonbonded settings
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
DispCorr = EnerPres ; account for cut-off vdW scheme
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling is on
pcoupl = Parrinello-Rahman ; Pressure coupling on in NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
refcoord_scaling = com
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Velocity generation
gen_vel = no ; Velocity generation is off
vim md.md
title = OPLS Lysozyme NPT equilibration
; Run parameters
integrator = md ; leap-frog integrator
nsteps = 500000 ; 2 * 500000 = 1000 ps (1 ns)
dt = 0.002 ; 2 fs
; Output control
nstxout = 0 ; suppress bulky .trr file by specifying
nstvout = 0 ; 0 for output frequency of nstxout,
nstfout = 0 ; nstvout, and nstfout
nstenergy = 5000 ; save energies every 10.0 ps
nstlog = 5000 ; update log file every 10.0 ps
nstxout-compressed = 5000 ; save compressed coordinates every 10.0 ps
compressed-x-grps = System ; save the whole system
; Bond parameters
continuation = yes ; Restarting after NPT
constraint_algorithm = lincs ; holonomic constraints
constraints = h-bonds ; bonds involving H are constrained
lincs_iter = 1 ; accuracy of LINCS
lincs_order = 4 ; also related to accuracy
; Neighborsearching
cutoff-scheme = Verlet ; Buffered neighbor searching
ns_type = grid ; search neighboring grid cells
nstlist = 10 ; 20 fs, largely irrelevant with Verlet scheme
rcoulomb = 1.0 ; short-range electrostatic cutoff (in nm)
rvdw = 1.0 ; short-range van der Waals cutoff (in nm)
; Electrostatics
coulombtype = PME ; Particle Mesh Ewald for long-range electrostatics
pme_order = 4 ; cubic interpolation
fourierspacing = 0.16 ; grid spacing for FFT
; Temperature coupling is on
tcoupl = V-rescale ; modified Berendsen thermostat
tc-grps = Protein Non-Protein ; two coupling groups - more accurate
tau_t = 0.1 0.1 ; time constant, in ps
ref_t = 300 300 ; reference temperature, one for each group, in K
; Pressure coupling is on
pcoupl = Parrinello-Rahman ; Pressure coupling on in NPT
pcoupltype = isotropic ; uniform scaling of box vectors
tau_p = 2.0 ; time constant, in ps
ref_p = 1.0 ; reference pressure, in bar
compressibility = 4.5e-5 ; isothermal compressibility of water, bar^-1
; Periodic boundary conditions
pbc = xyz ; 3-D PBC
; Dispersion correction
DispCorr = EnerPres ; account for cut-off vdW scheme
; Velocity generation
gen_vel = no ; Velocity generation is off
2. Étapes de simulation formelle
Cible:Construisez un système de simulation physiquement raisonnable pour jeter les bases d’une simulation dynamique ultérieure.
1. Charger la structure moléculaire (fichier PDB):
• Fichiers PDB(Protein Data Bank) contient des coordonnées tridimensionnelles et des informations structurelles sur les molécules (telles que les protéines, les acides nucléiques et les petites molécules).
• GROMACS utilise l'outil pdb2gmx pour convertir ces informations de coordonnées en un modèle moléculaire et attribuer des paramètres de champ de force.
• Champ de force:Un modèle mathématique qui décrit les interactions entre les molécules, y compris les liaisons, les angles, l'énergie potentielle dièdre, les forces de van der Waals et les interactions de charge.
• Champs de force communs : GROMOS, AMBER, CHARMM.
(Les plus couramment utilisés sont la protéine AMBER99SB, le champ de force tout-atome OPLS-AA/L, le champ de force charm36 (vous devez le télécharger et le configurer vous-même), les autres champs de force sont trop anciens et ne conviennent pas à la publication d'articles)
2. Générer un fichier de topologie:
• Le fichier de topologie (topol.top) définit les paramètres du champ de force de chaque molécule dans le système de simulation, tels que le type d'atome, le type de liaison et ses paramètres, etc.
• Il se combine avec le fichier de coordonnées pour former la base des simulations de dynamique moléculaire.
3. Exécutez pdb2gmx, sélectionnez le champ de force : 15 et appuyez sur Entrée
#删除水分子(PDB 文件中的 “HOH” 残基)
grep -v HOH 1aki.pdb > 1AKI_clean.pdb
gmx_mpi pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce
#选择 15,按回车,OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce
:-) GROMACS - gmx pdb2gmx, 2023 (-:
Executable: /data/app/gromacs/bin/gmx_mpi
Data prefix: /data/app/gromacs
Working dir: /input0
Command line:
gmx_mpi pdb2gmx -f 1AKI_clean.pdb -o 1AKI_processed.gro -water spce
Select the Force Field:
From '/data/app/gromacs/share/gromacs/top':
1: AMBER03 protein, nucleic AMBER94 (Duan et al., J. Comp. Chem. 24, 1999-2012, 2003)
2: AMBER94 force field (Cornell et al., JACS 117, 5179-5197, 1995)
3: AMBER96 protein, nucleic AMBER94 (Kollman et al., Acc. Chem. Res. 29, 461-469, 1996)
4: AMBER99 protein, nucleic AMBER94 (Wang et al., J. Comp. Chem. 21, 1049-1074, 2000)
5: AMBER99SB protein, nucleic AMBER94 (Hornak et al., Proteins 65, 712-725, 2006)
6: AMBER99SB-ILDN protein, nucleic AMBER94 (Lindorff-Larsen et al., Proteins 78, 1950-58, 2010)
7: AMBERGS force field (Garcia & Sanbonmatsu, PNAS 99, 2782-2787, 2002)
8: CHARMM27 all-atom force field (CHARM22 plus CMAP for proteins)
9: GROMOS96 43a1 force field
10: GROMOS96 43a2 force field (improved alkane dihedrals)
11: GROMOS96 45a3 force field (Schuler JCC 2001 22 1205)
12: GROMOS96 53a5 force field (JCC 2004 vol 25 pag 1656)
13: GROMOS96 53a6 force field (JCC 2004 vol 25 pag 1656)
14: GROMOS96 54a7 force field (Eur. Biophys. J. (2011), 40,, 843-856, DOI: 10.1007/s00249-011-0700-9)
15: OPLS-AA/L all-atom force field (2001 aminoacid dihedrals)
15
Using the Oplsaa force field in directory oplsaa.ff
going to rename oplsaa.ff/aminoacids.r2b
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.r2b
Reading 1AKI_clean.pdb...
WARNING: all CONECT records are ignored
Read 'LYSOZYME', 1001 atoms
Analyzing pdb file
Splitting chemical chains based on TER records or chain id changing.
There are 1 chains and 0 blocks of water and 129 residues with 1001 atoms
chain #res #atoms
1 'A' 129 1001
All occupancies are one
All occupancies are one
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/atomtypes.atp
Reading residue database... (Oplsaa)
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.rtp
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.hdb
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.n.tdb
Opening force field file /data/app/gromacs/share/gromacs/top/oplsaa.ff/aminoacids.c.tdb
Processing chain 1 'A' (1001 atoms, 129 residues)
Analysing hydrogen-bonding network for automated assignment of histidine
protonation. 213 donors and 184 acceptors were found.
There are 255 hydrogen bonds
Will use HISE for residue 15
Identified residue LYS1 as a starting terminus.
Identified residue LEU129 as a ending terminus.
8 out of 8 lines of specbond.dat converted successfully
Special Atom Distance matrix:
CYS6 MET12 HIS15 CYS30 CYS64 CYS76 CYS80
SG48 SD87 NE2118 SG238 SG513 SG601 SG630
MET12 SD87 1.166
HIS15 NE2118 1.776 1.019
CYS30 SG238 1.406 1.054 2.069
CYS64 SG513 2.835 1.794 1.789 2.241
CYS76 SG601 2.704 1.551 1.468 2.116 0.765
CYS80 SG630 2.959 1.951 1.916 2.391 0.199 0.944
CYS94 SG724 2.550 1.407 1.382 1.975 0.665 0.202 0.855
MET105 SD799 1.827 0.911 1.683 0.888 1.849 1.461 2.036
CYS115 SG889 1.576 1.084 2.078 0.200 2.111 1.989 2.262
CYS127 SG981 0.197 1.072 1.721 1.313 2.799 2.622 2.934
CYS94 MET105 CYS115
SG724 SD799 SG889
MET105 SD799 1.381
CYS115 SG889 1.853 0.790
CYS127 SG981 2.475 1.686 1.483
Linking CYS-6 SG-48 and CYS-127 SG-981...
Linking CYS-30 SG-238 and CYS-115 SG-889...
Linking CYS-64 SG-513 and CYS-80 SG-630...
Linking CYS-76 SG-601 and CYS-94 SG-724...
Start terminus LYS-1: NH3+
End terminus LEU-129: COO-
Checking for duplicate atoms....
Generating any missing hydrogen atoms and/or adding termini.
Now there are 129 residues with 1960 atoms
Making bonds...
Number of bonds was 1984, now 1984
Generating angles, dihedrals and pairs...
Before cleaning: 5142 pairs
Before cleaning: 5187 dihedrals
Making cmap torsions...
There are 5187 dihedrals, 426 impropers, 3547 angles
5106 pairs, 1984 bonds and 0 virtual sites
Total mass 14313.193 a.m.u.
Total charge 8.000 e
Writing topology
Writing coordinate file...
--------- PLEASE NOTE ------------
You have successfully generated a topology from: 1AKI_clean.pdb.
The Oplsaa force field and the spce water model are used.
--------- ETON ESAELP ------------
GROMACS reminds you: "Any one who considers arithmetical methods of producing random digits is, of course, in a state of sin." (John von Neumann)
Vous pouvez voir qu'il y a plusieurs autres fichiers, et « Charge totale 8 000 e ». Nous devrons neutraliser la charge plus tard
Utilisez la commande vim pour afficher topol.top et vous pourrez voir la balise du champ de force.
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# ls
1AKI_clean.pdb 1aki.pdb md.mdp npt.mdp posre.itp
1AKI_processed.gro ions.mdp minim.mdp nvt.mdp topol.top
4. Créez une boîte en dodécaèdre rhombique pour enfermer la protéine afin que nous puissions ajouter des molécules de solvant
• Sélection de la forme de la boîte:Les formes de boîte de simulation courantes incluent le cube, la boîte orthogonale, le dodécaèdre rhombique, etc. Les formes qui peuvent remplir efficacement les molécules et réduire la quantité de calcul sont généralement choisies.
gmx_mpi editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
#将蛋白质在框中居中(-c),并将其放置在框边缘至少 1.0 nm 的位置(-d 1.0)
Command line:
gmx_mpi editconf -f 1AKI_processed.gro -o 1AKI_newbox.gro -c -d 1.0 -bt cubic
Note that major changes are planned in future for editconf, to improve usability and utility.
Read 1960 atoms
Volume: 123.376 nm^3, corresponds to roughly 55500 electrons
No velocities found
system size : 3.817 4.234 3.454 (nm)
diameter : 5.010 (nm)
center : 2.781 2.488 0.017 (nm)
box vectors : 5.906 6.845 3.052 (nm)
box angles : 90.00 90.00 90.00 (degrees)
box volume : 123.38 (nm^3)
shift : 0.724 1.017 3.488 (nm)
new center : 3.505 3.505 3.505 (nm)
new box vectors : 7.010 7.010 7.010 (nm)
new box angles : 90.00 90.00 90.00 (degrees)
new box volume : 344.48 (nm^3)
5. Définissez une boîte et ajoutez du solvant (eau)
But:Solvatation:Placez les molécules dans un environnement solvant (comme une boîte à eau) pour simuler leur comportement dans l'environnement réel.
gmx_mpi solvate -cp 1AKI_newbox.gro -cs spc216.gro -o 1AKI_solv.gro -p topol.top
#结果
Generating solvent configuration
Will generate new solvent configuration of 4x4x4 boxes
Solvent box contains 39252 atoms in 13084 residues
Removed 5451 solvent atoms due to solvent-solvent overlap
Removed 1869 solvent atoms due to solute-solvent overlap
Sorting configuration
Found 1 molecule type:
SOL ( 3 atoms): 10644 residues
Generated solvent containing 31932 atoms in 10644 residues
Writing generated configuration to 1AKI_solv.gro
Output configuration contains 33892 atoms in 10773 residues
Volume : 344.484 (nm^3)
Density : 997.935 (g/l)
Number of solvent molecules: 10644
Processing topology
Adding line for 10644 solvent molecules with resname (SOL) to topology file (topol.top)
#查看一下 top 文件
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# ls
'#topol.top.1#' 1AKI_newbox.gro 1AKI_solv.gro ions.mdp minim.mdp nvt.mdp topol.top
1AKI_clean.pdb 1AKI_processed.gro 1aki.pdb md.mdp npt.mdp posre.itp
(base) root@liangzhong-4ay9ej85pxvd-main:/input0# vim topol.top
#查看 top 文件的末尾
Vous pouvez voir [des molécules]
protein_chain_A, chaîne de protéine A
Molécule d'eau SOL
6. Assemblez le fichier .tpr
gmx_mpi grompp -f ions.mdp -c 1AKI_solv.gro -p topol.top -o ions.tpr
7. Neutraliser la charge et remplacer les molécules de solvant par des ions
Neutralisation de charge:Ajoutez des ions (tels que Na⁺, Cl⁻) pour neutraliser la charge totale du système et éviter la distorsion de simulation causée par les effets électrostatiques.
gmx_mpi genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
Command line:
gmx_mpi genion -s ions.tpr -o 1AKI_solv_ions.gro -p topol.top -pname NA -nname CL -neutral
Reading file ions.tpr, VERSION 2023 (single precision)
Reading file ions.tpr, VERSION 2023 (single precision)
Will try to add 0 NA ions and 8 CL ions.
Select a continuous group of solvent molecules
Group 0 ( System) has 33892 elements
Group 1 ( Protein) has 1960 elements
Group 2 ( Protein-H) has 1001 elements
Group 3 ( C-alpha) has 129 elements
Group 4 ( Backbone) has 387 elements
Group 5 ( MainChain) has 517 elements
Group 6 ( MainChain+Cb) has 634 elements
Group 7 ( MainChain+H) has 646 elements
Group 8 ( SideChain) has 1314 elements
Group 9 ( SideChain-H) has 484 elements
Group 10 ( Prot-Masses) has 1960 elements
Group 11 ( non-Protein) has 31932 elements
Group 12 ( Water) has 31932 elements
Group 13 ( SOL) has 31932 elements
Group 14 ( non-Water) has 1960 elements
Select a group: 13
Selected 13: 'SOL'
Number of (3-atomic) solvent molecules: 10644
Processing topology
Replacing 8 solute molecules in topology file (topol.top) by 0 NA and 8 CL ions.
Back Off! I just backed up topol.top to ./#topol.top.2#
Using random seed -1212354563.
Replacing solvent molecule 3671 (atom 12973) with CL
Replacing solvent molecule 2264 (atom 8752) with CL
Replacing solvent molecule 2559 (atom 9637) with CL
Replacing solvent molecule 8081 (atom 26203) with CL
Replacing solvent molecule 8468 (atom 27364) with CL
Replacing solvent molecule 7439 (atom 24277) with CL
Replacing solvent molecule 9983 (atom 31909) with CL
Replacing solvent molecule 650 (atom 3910) with CL
GROMACS reminds you: "Water is just water" (Berk Hess)
(base) root@liangzhong-4ay9ej85pxvd-main:/input0#
On peut voir que beaucoup d’ions chlorure ont été ajoutés
8. Minimisation de l'énergie
8.1 Raison : Lorsque nous effectuons une simulation de dynamique moléculaire, les fichiers pdb que nous obtenons sont obtenus par rayons X, microscopie électronique et autres méthodes, qui contiennent de nombreux angles de liaison et distorsions avec une énergie excessive, ou la formation ou la rupture de liaisons hydrogène, des arrangements spéciaux entre les molécules et une énergie élevée causée par la faible distance entre les atomes, ce qui rend difficile pour le système moléculaire dans la simulation de passer d'un état de haute énergie à un état de basse énergie. Cette méthode est nécessaire pour minimiser l’énergie du système.
8.2 Principe : Le principe de base de la minimisation de l'énergie : La minimisation de l'énergie est basée sur la fonction d'énergie potentielle, qui réduit l'énergie potentielle totale du système en calculant et en ajustant de manière itérative les coordonnées atomiques. Les méthodes couramment utilisées comprennent :
1 Méthode du gradient conjugué : une méthode de descente de gradient qui accélère la convergence en utilisant les informations de l'étape précédente.
2 Méthode de descente la plus raide : chaque itération se déplace dans la direction opposée du gradient d'énergie potentielle, réduisant l'énergie via le chemin de descente le plus raide.
③ Méthode de Newton-Raphson : une méthode de dérivée du second ordre qui utilise la dérivée du second ordre de la fonction d'énergie potentielle pour trouver plus précisément le point d'énergie minimum.
8.3 Objectif : Trouver une conformation moléculaire plus stable en ajustant les coordonnées atomiques de la molécule pour réduire l'énergie potentielle totale du système.
1. Supprimer les conformations géométriques déraisonnables : Grâce à la minimisation de l'énergie, les conformations géométriques déraisonnables dans la structure initiale, telles que le chevauchement et l'étirement atomiques déraisonnables, peuvent être supprimées, l'état d'énergie élevé qu'elles provoquent peut être éliminé et le système peut être rendu plus stable.
2. Préparez la structure initiale : assurez-vous que la structure initiale est dans un état d'énergie potentielle faible pour éviter un impact énergétique élevé inutile pendant la simulation.
③ Améliorer l'efficacité du calcul : en réduisant l'énergie potentielle totale du système, la stabilité et l'efficacité des simulations et des calculs ultérieurs peuvent être améliorées
gmx_mpi grompp -f minim.mdp -c 1AKI_solv_ions.gro -p topol.top -o em.tpr
gmx_mpi mdrun -v -deffnm em

9. Regardez les résultats (à la fin du tutoriel, nous vous apprendrons à visualiser des fichiers xvg. Vous pouvez utiliser xmgrace, qtgrace, Python et Excel pour créer un graphique. Il s'agit essentiellement d'un graphique linéaire).
gmx_mpi energy -f em.edr -o potential.xvg
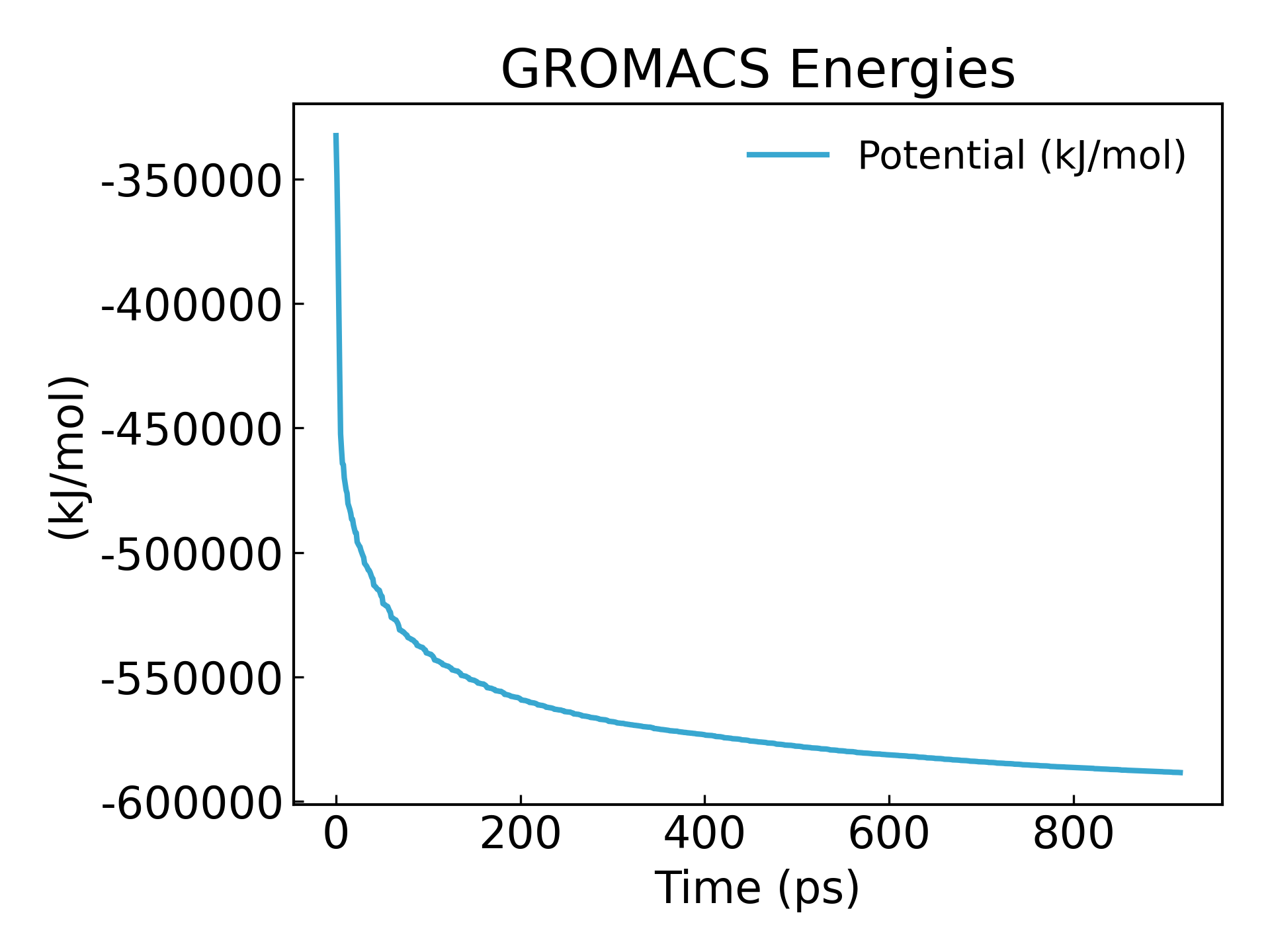
On peut voir que l’énergie est minimisée à -600 000 kj/mol
10. Équilibre du système
Objectif : Ajuster le système aux conditions de température et de pression cibles pour le rendre proche de l'état physique réel.
(1) Simulation NVT (volume constant et température constante) :
• Un ensemble à volume constant et à température constante est utilisé pour stabiliser la température du système à une valeur cible.
• Contrôleur de température : Les coupleurs de température Berendsen ou V-rescale (méthode de couplage faible modifiée) couramment utilisés contrôlent la température en ajustant la vitesse atomique.
(2) Simulation NPT (pression constante et température constante) :
• L'ensemble pression constante et température constante ajuste en outre la densité du système à une valeur cible (généralement la densité de l'eau liquide ~1 g/cm³).
• Régulateur de pression : Les coupleurs de pression Berendsen ou la méthode de contrôle de pression Parrinello-Rahman sont couramment utilisés.
10.1. Effectuer l'équilibrage NVT pendant 100 ps, qui est effectué sous (nombre de particules, volume et température constants), également appelé « isotherme et isochore »
Il est très rapide avec l'accélération GPU, ne prenant que 10+ secondes.
gmx_mpi grompp -f nvt.mdp -c em.gro -r em.gro -p topol.top -o nvt.tpr
gmx_mpi mdrun -deffnm nvt -nb gpu -pme cpu
#将 PME 任务移至 CPU

gmx_mpi energy -f nvt.edr -o temperature.xvg
#生成温度随时间变化的图像,查看温度是否平衡
16
0
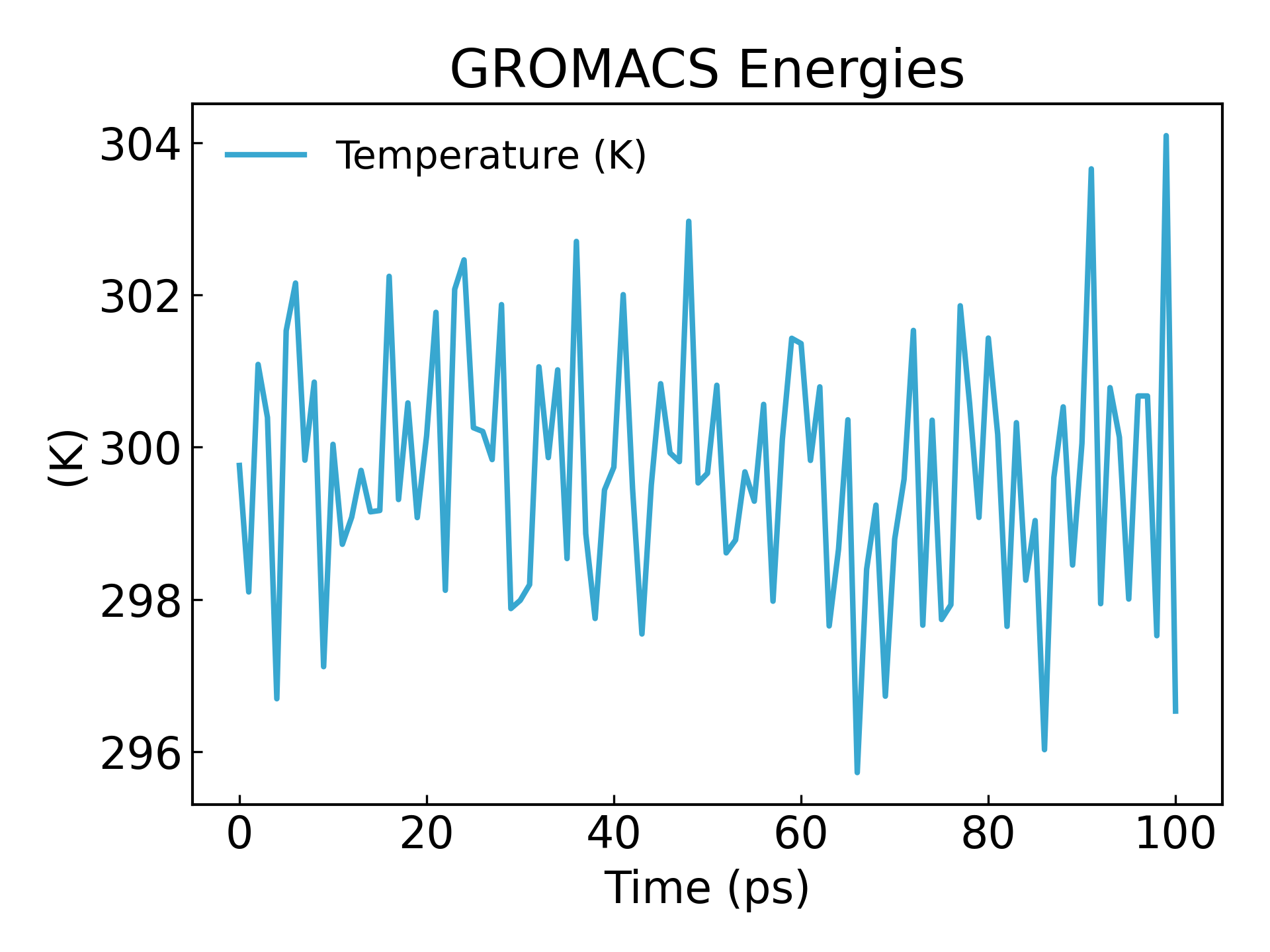
On peut voir que la température atteint également un état stable de base en 100 ps
10.2. Équilibrage NPT « isotherme et isobare », stabilise la pression du système et effectue un équilibrage NPT de 100 ps.
gmx_mpi grompp -f npt.mdp -c nvt.gro -r nvt.gro -t nvt.cpt -p topol.top -o npt.tpr
gmx_mpi mdrun -deffnm npt -nb gpu -pme cpu
#压力是否平衡
gmx_mpi energy -f npt.edr -o pressure.xvg
18
0
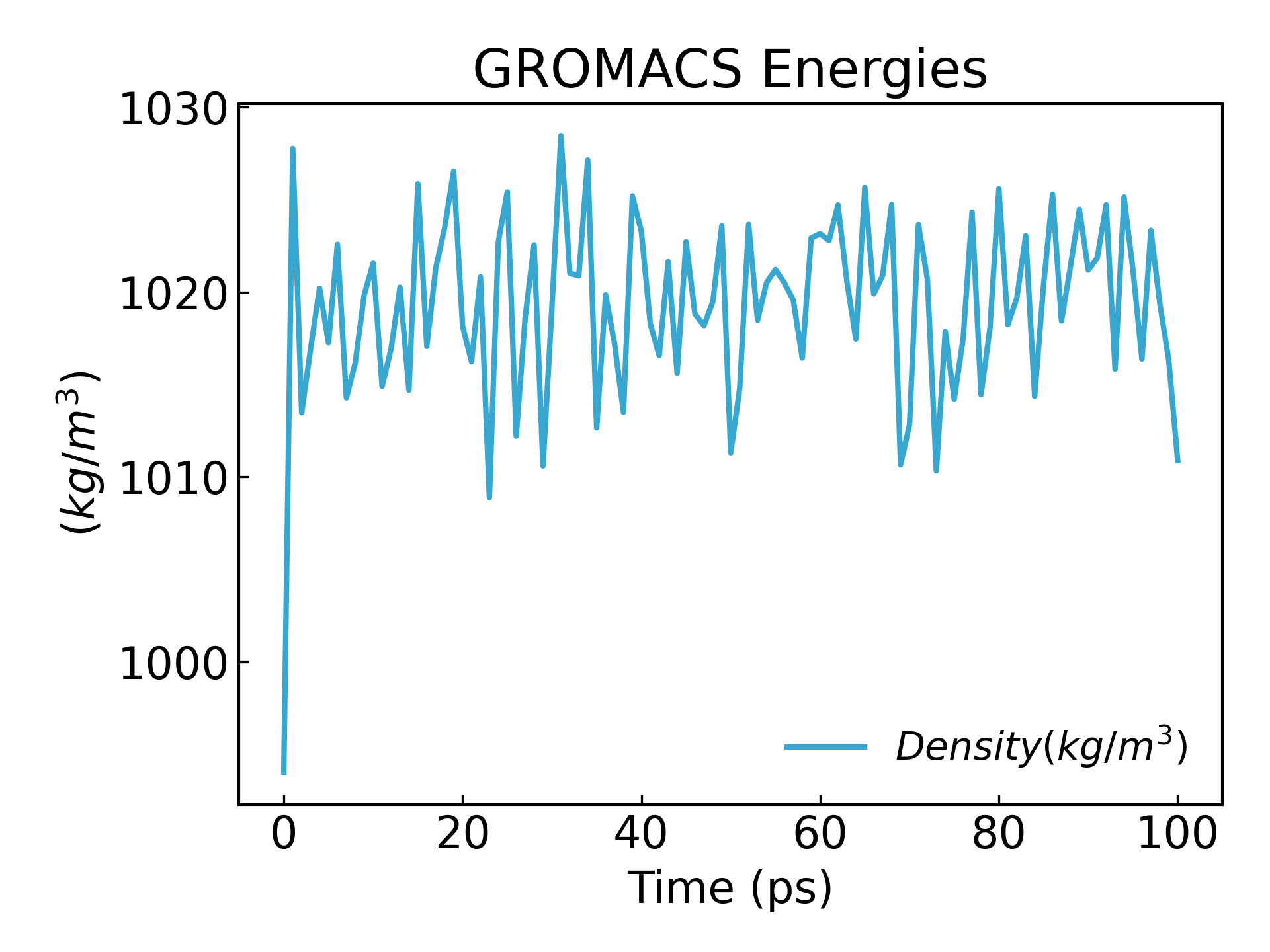
gmx_mpi energy -f npt.edr -o density.xvg
24
0
Nous pouvons voir que la densité atteint un état stationnaire :
11. Après avoir terminé les 2 étapes d’équilibrage, le système est maintenant bien équilibré à la température et à la pression souhaitées. Nous pouvons maintenant libérer les contraintes de position et exécuter MD pour continuer.
Vous pouvez modifier l'heure selon vos besoins. Ce tutoriel simule 100 ns.
Pas de temps dt = 2 fs (réglage courant) :
50000000 pas, avec un pas de temps de 2 fs, équivalent à 100 nanosecondes (ns)
50000000 x2 fs = 10^8 fs = 10^5 ps = 100ns
vim md.mdp
nsteps = 50000000 ; 2 * 50000000 = 100000 ps (100 ns)

gmx_mpi grompp -f md.mdp -c npt.gro -t npt.cpt -p topol.top -o md_0_1.tpr
##最后一步提交,pme 分配到 CPU 上:
gmx_mpi mdrun -deffnm md_0_1 -v -nb gpu -pme cpu
-v peut afficher le temps restant de l'exécution.
3. Analyse des résultats
1. trjconv, qui est utilisé comme outil de post-traitement pour supprimer des coordonnées, corriger la périodicité ou modifier manuellement la trajectoire (unité de temps, fréquence d'images, etc.). Cela est dû au fait que dans toutes les simulations avec des conditions aux limites périodiques, les molécules peuvent se briser ou sauter autour du bord de la boîte. Il peut recentrer la molécule dans la boîte, réenvelopper la molécule et restaurer la forme dodécaédrique rhombique de la boîte.
-pbc mol: 去除轨迹中的周期性边界条件 (PBC),并基于分子进行修正(即确保每个分子在轨迹中是完整的)。
• -center: 将选定的分子/分组居中到模拟框的中心。
gmx_mpi trjconv -s md_0_1.tpr -f md_0_1.xtc -o md_0_1_noPBC.xtc -pbc mol -center
Select group for centering
Group 0 ( System) has 33876 elements
Group 1 ( Protein) has 1960 elements
Group 2 ( Protein-H) has 1001 elements
Group 3 ( C-alpha) has 129 elements
Group 4 ( Backbone) has 387 elements
Group 5 ( MainChain) has 517 elements
Group 6 ( MainChain+Cb) has 634 elements
Group 7 ( MainChain+H) has 646 elements
Group 8 ( SideChain) has 1314 elements
Group 9 ( SideChain-H) has 484 elements
Group 10 ( Prot-Masses) has 1960 elements
Group 11 ( non-Protein) has 31916 elements
Group 12 ( Water) has 31908 elements
Group 13 ( SOL) has 31908 elements
Group 14 ( non-Water) has 1968 elements
Group 15 ( Ion) has 8 elements
Group 16 ( Water_and_ions) has 31916 elements
Select a group: 1
Selected 1: 'Protein'
Select group for output
Group 0 ( System) has 33876 elements
Group 1 ( Protein) has 1960 elements
Group 2 ( Protein-H) has 1001 elements
Group 3 ( C-alpha) has 129 elements
Group 4 ( Backbone) has 387 elements
Group 5 ( MainChain) has 517 elements
Group 6 ( MainChain+Cb) has 634 elements
Group 7 ( MainChain+H) has 646 elements
Group 8 ( SideChain) has 1314 elements
Group 9 ( SideChain-H) has 484 elements
Group 10 ( Prot-Masses) has 1960 elements
Group 11 ( non-Protein) has 31916 elements
Group 12 ( Water) has 31908 elements
Group 13 ( SOL) has 31908 elements
Group 14 ( non-Water) has 1968 elements
Group 15 ( Ion) has 8 elements
Group 16 ( Water_and_ions) has 31916 elements
Select a group: 0
Selected 0: 'System'
La sélection 4 (« Backbone ») a été utilisée pour l’ajustement par les moindres carrés et les calculs RMSD.
2.RMSD
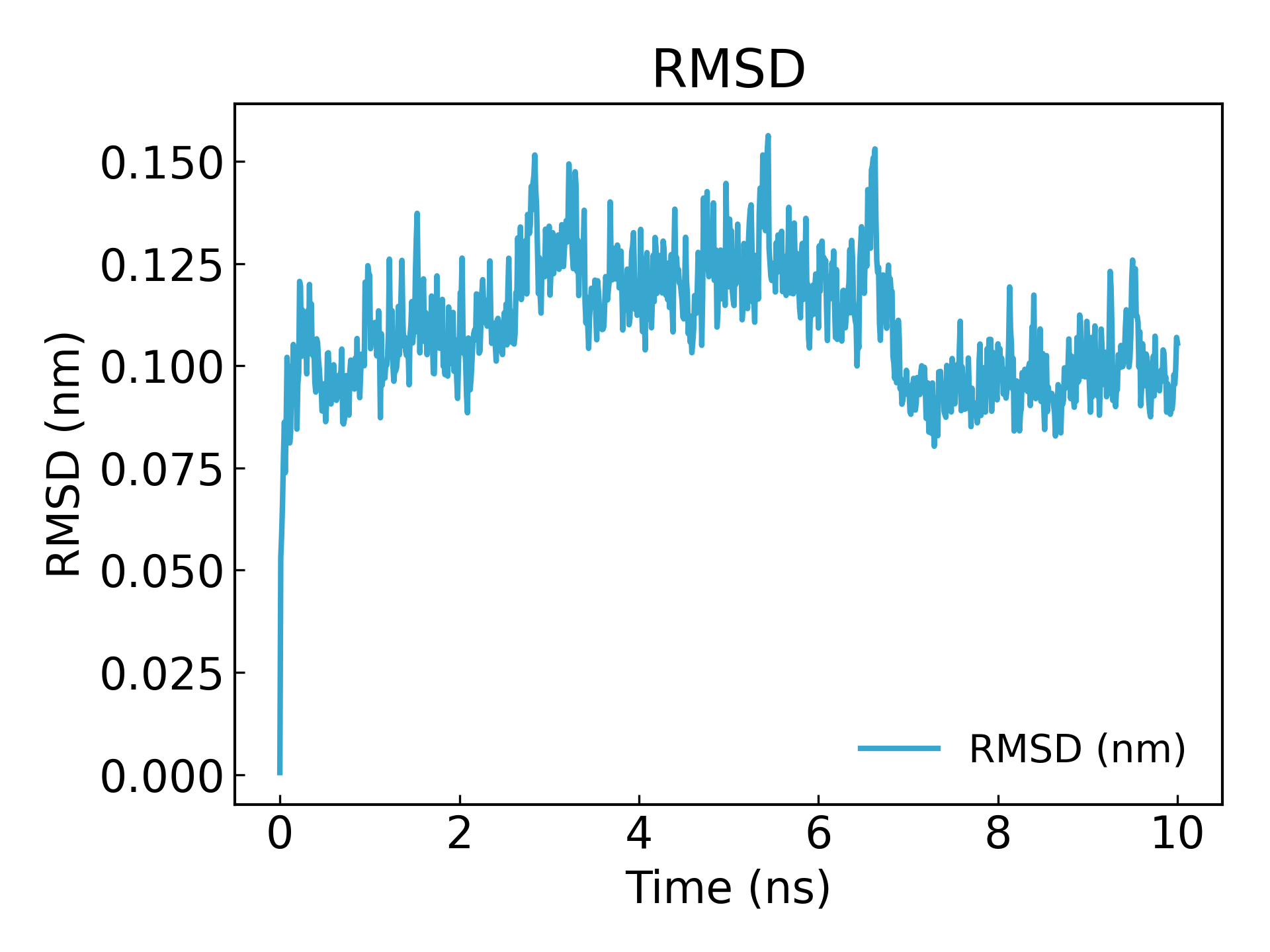
Peut être utilisé pour vérifier la convergence de la simulation et la stabilité de la protéine.g_rms**Le RMSD de la structure pendant la simulation et la structure initiale, l'écart de la structure à un certain moment par rapport à 0 ns, est souvent utilisé pour évaluer la stabilité de la structure des protéines. Il est souvent utilisé dans le domaine de la conception de médicaments, comme la stabilité de la structure des ligands protéiques. La plage de fluctuation de la courbe RMSD est faible et stable, ce qui indique que l'affinité entre le ligand et le récepteur est grande.
gmx_mpi rms -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rmsd.xvg
Sélectionnez 4 (« Backbone ») pour l'ajustement par les moindres carrés et le calcul de la RMSD
On peut voir que l'équilibre est atteint après 10 ns (Remarque : lors de la publication ou de la soumission d'un article, il est préférable de simuler pendant 100 ns ou 50 ns)
3. Analyse du rayon de giration
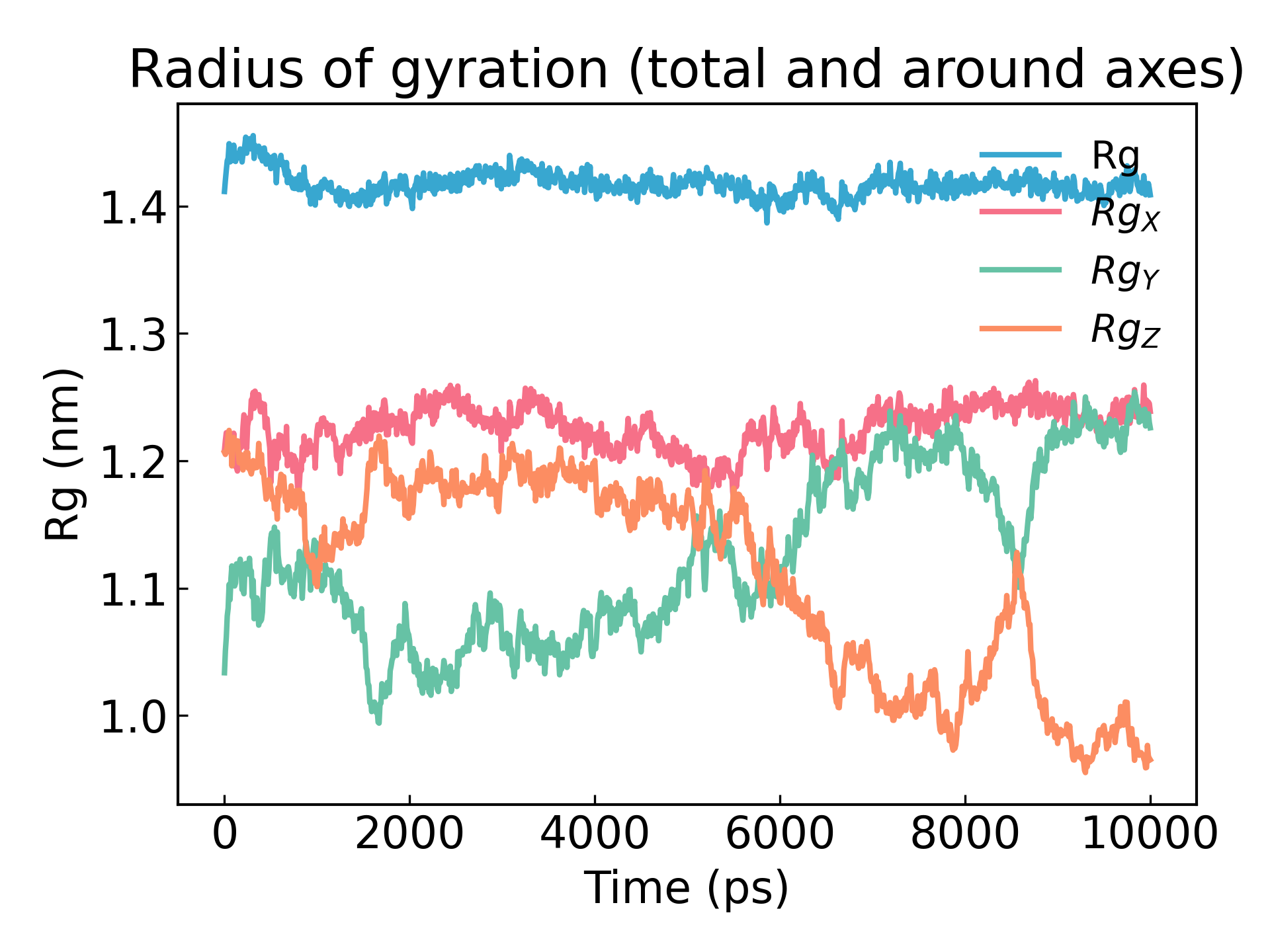
gmx_mpi gyrate -s md_0_1.tpr -f md_0_1_noPBC.xtc -o gyrate.xvg
Le rayon de giration d’une protéine est une mesure de sa compacité. Si une structure protéique est stable, elle conservera probablement une valeur R g relativement stable. Si une protéine se déplie, sa valeur R g changera avec le temps. Analysons le rayon de giration du lysozyme dans notre simulation :
4. Visualisation
Logiciels recommandés :DuIvyTools : outils d'analyse et de visualisation de simulation GROMACS
Depuis:https://github.com/CharlesHahn/DuIvyTools
pip install DuIvyTools
dit xvg_show -f rmsd.xvg -o rmsd_plot.png
dit xvg_show -f rmsd.xvg -o rmsd_plot.png
dit xvg_show -f potential.xvg -o potential_plot.png
dit xvg_show -f temperature.xvg -o temperature_plot.png
dit xvg_show -f density.xvg -o density_plot.png
dit xvg_show -f pressure.xvg -o pressure_plot.png
Ouvrez l'ensemble de données que nous avons créé
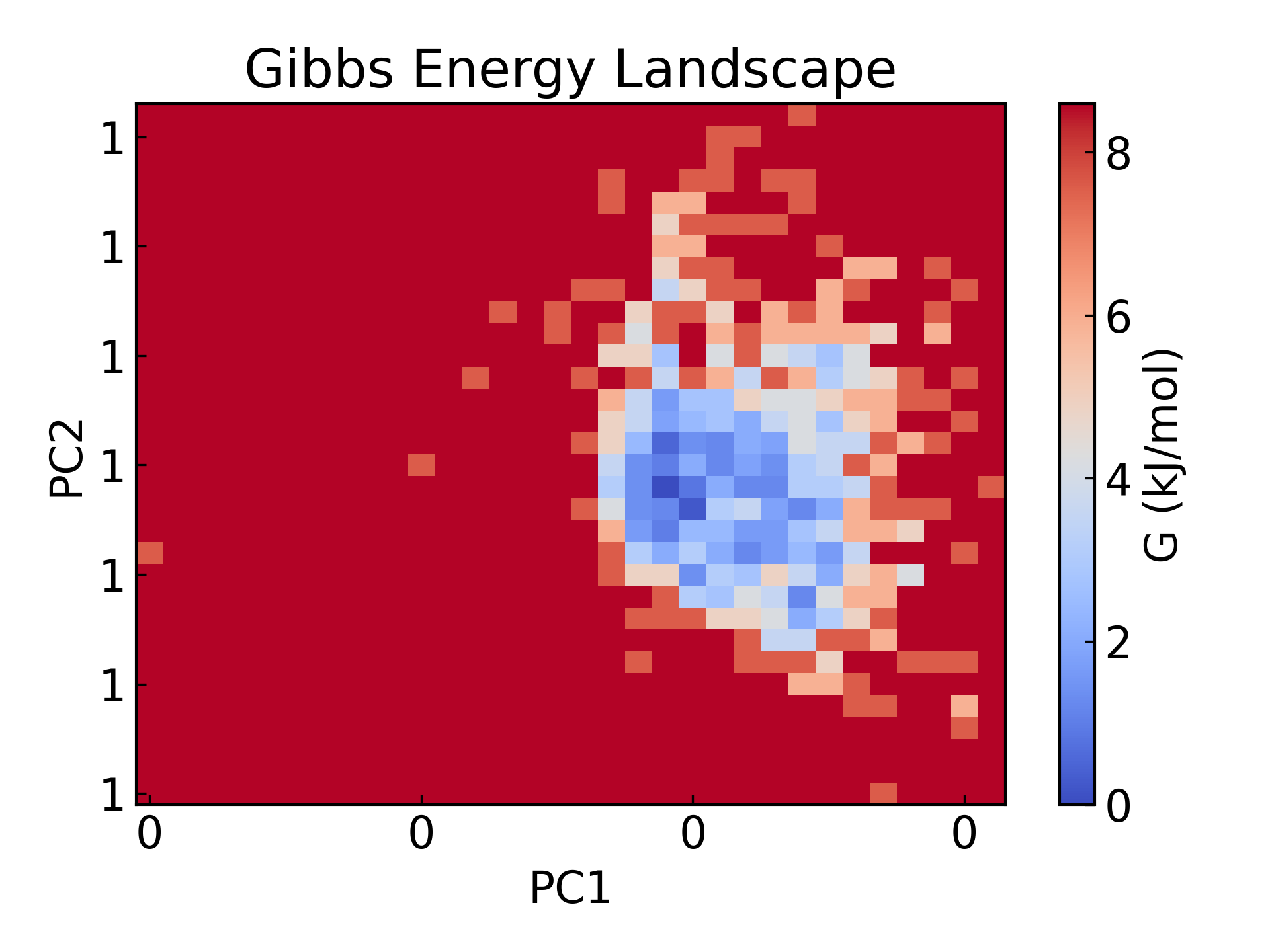
5. Cartographie du paysage énergétique GROMACS Le paysage énergétique libre a été tracé, avec le rayon de giration et le rmsd comme deux composants PCA respectivement.
gmx_mpi gyrate -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rg.xvg
gmx_mpi rms -s md_0_1.tpr -f md_0_1_noPBC.xtc -o rmsd.xvg
vim rmsd.xvg
#输入以下命令删除以 # 或 @ 开头的行:
:g/^[@#]/d
vim rg.xvg
#输入以下命令删除以 # 或 @ 开头的行:
:g/^[@#]/d
#注意不要有空行,
paste rmsd.xvg rg.xvg > rmsd-rg.xvg
(base) # tail -f rmsd-rg.xvg
9910.0000000 0.0925907 9910 1.37624 1.20925 1.20765 0.931324
9920.0000000 0.0881348 9920 1.38369 1.22248 1.21078 0.932077
9930.0000000 0.0911074 9930 1.39224 1.23799 1.21709 0.928842
9940.0000000 0.0893596 9940 1.38188 1.21942 1.21672 0.922916
9950.0000000 0.0915931 9950 1.37509 1.21939 1.20194 0.922051
9960.0000000 0.0978161 9960 1.38113 1.2262 1.21084 0.919414
9970.0000000 0.0954911 9970 1.37934 1.21241 1.20711 0.937075
9980.0000000 0.0993617 9980 1.38301 1.22353 1.21083 0.92862
9990.0000000 0.1069279 9990 1.37943 1.22317 1.20579 0.924978
10000.0000000 0.1055321 10000 1.37524 1.21648 1.20194 0.92632
^Z
[11]+ Stopped tail -f rmsd-rg.xvg
#查看已经将两个文件整合在一起了,我们只要保留
#从 rmsd-rg.xvg 文件内容可以看到,每一行的数据格式如下:
时间_RMSD(ns) RMSD 时间_Rg(ps) Rg 其他列
(base) tail -f rmsd.xvg
9910.0000000 0.0925907
9920.0000000 0.0881348
9930.0000000 0.0911074
9940.0000000 0.0893596
9950.0000000 0.0915931
9960.0000000 0.0978161
9970.0000000 0.0954911
9980.0000000 0.0993617
9990.0000000 0.1069279
10000.0000000 0.1055321
^Z
[12]+ Stopped tail -f rmsd.xvg
#整理数据为
时间 (ns) RMSD Rg
(base) python
Python 3.8.15 (default, Nov 24 2022, 15:19:38)
[GCC 11.2.0] :: Anaconda, Inc. on linux
Type "help", "copyright", "credits" or "license" for more information.
# 加载数据
>>> import pandas as pd
>>> data = pd.read_csv("rmsd-rg.xvg", delim_whitespace=True, header=None, comment="#")
# 保留需要的列:第 1 列 (RMSD 时间) 、第 2 列 (RMSD) 、第 4 列 (Rg)
>>> cleaned_data = data[[0, 1, 3]]
# 将列名修改为更直观的名称
>>> cleaned_data.columns = ["Time (ps)", "RMSD", "Rg"]
>>> cleaned_data.to_csv("rmsd-rg-cleaned.xvg", sep="\t", index=False, header=False)
>>> exit()
#整理成功
tail -f rmsd-rg-cleaned.xvg
9910.0 0.0925907 1.37624
9920.0 0.0881348 1.38369
9930.0 0.0911074 1.39224
9940.0 0.0893596 1.38188
9950.0 0.0915931 1.37509
9960.0 0.0978161 1.38113
9970.0 0.0954911 1.37934
9980.0 0.0993617 1.38301
9990.0 0.1069279 1.37943
10000.0 0.1055321 1.37524
gmx_mpi sham -tsham 300 -nlevels 100 -f rmsd-rg-cleaned.xvg -ls gibbs.xpm -g gibbs.log -lsh enthalpy.xpm -lss entropy.xpm
dit xpm_show -f gibbs.xpm -o gibbs_2d.png
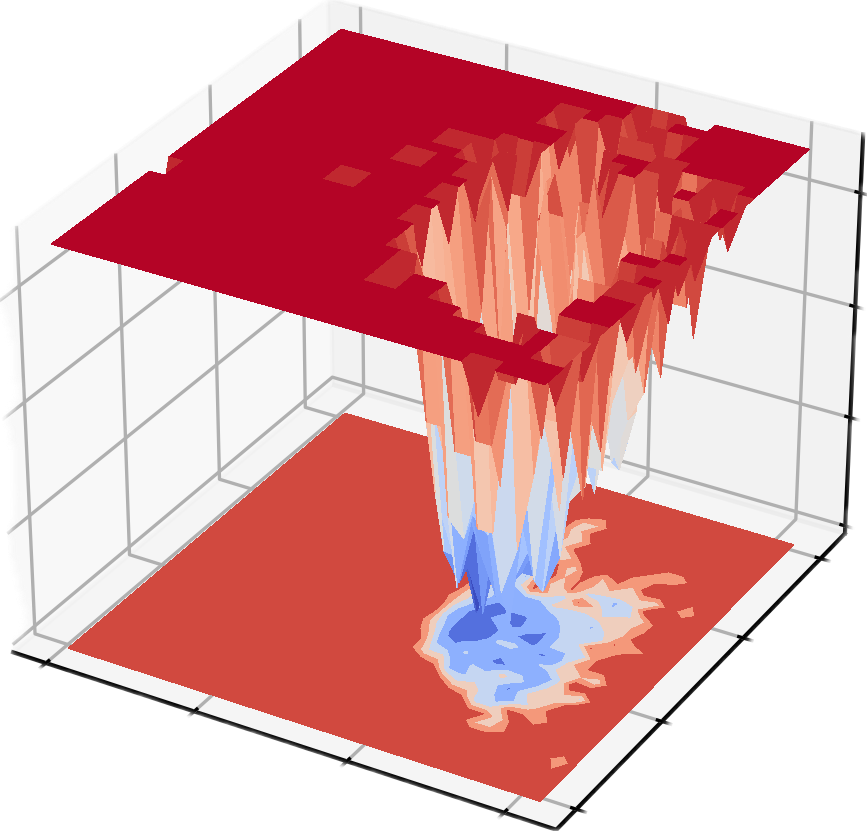
dit xpm_show -f gibbs.xpm -m 3d -o gibbs_3d.png
#tsham : 设定温度
#-nlevels: 设定 FEL 的层次数量
Free Energy Landscape (FES) est un outil très important dans la simulation moléculaire, qui est principalement utilisé pour décrire la distribution d'énergie libre des systèmes moléculaires à des coordonnées spécifiques. Sa fonction principale est de révéler la stabilité thermodynamique et le processus cinétique du système, ce qui est généralement d'une grande importance dans les domaines de recherche scientifique suivants :
- Études à l'état stationnaire et à l'état de transition:Révéler la conformation stable (point minimum d'énergie libre) et le chemin de conversion conformationnelle (barrière d'énergie libre) du système, qui est utilisé pour analyser le repliement des protéines, les réactions chimiques et les processus de reconnaissance moléculaire.
- Voies cinétiques et stabilité thermodynamique:La quantification de la différence d’énergie libre entre différents états permet de comprendre la force motrice énergétique des interactions moléculaires et des transitions conformationnelles.
- Recherche sur la conception de médicaments et la catalyse:Prédire les voies de liaison ligand-récepteur, les barrières énergétiques et les taux de réaction, fournissant des conseils théoriques pour l'analyse des mécanismes moléculaires et l'optimisation fonctionnelle.
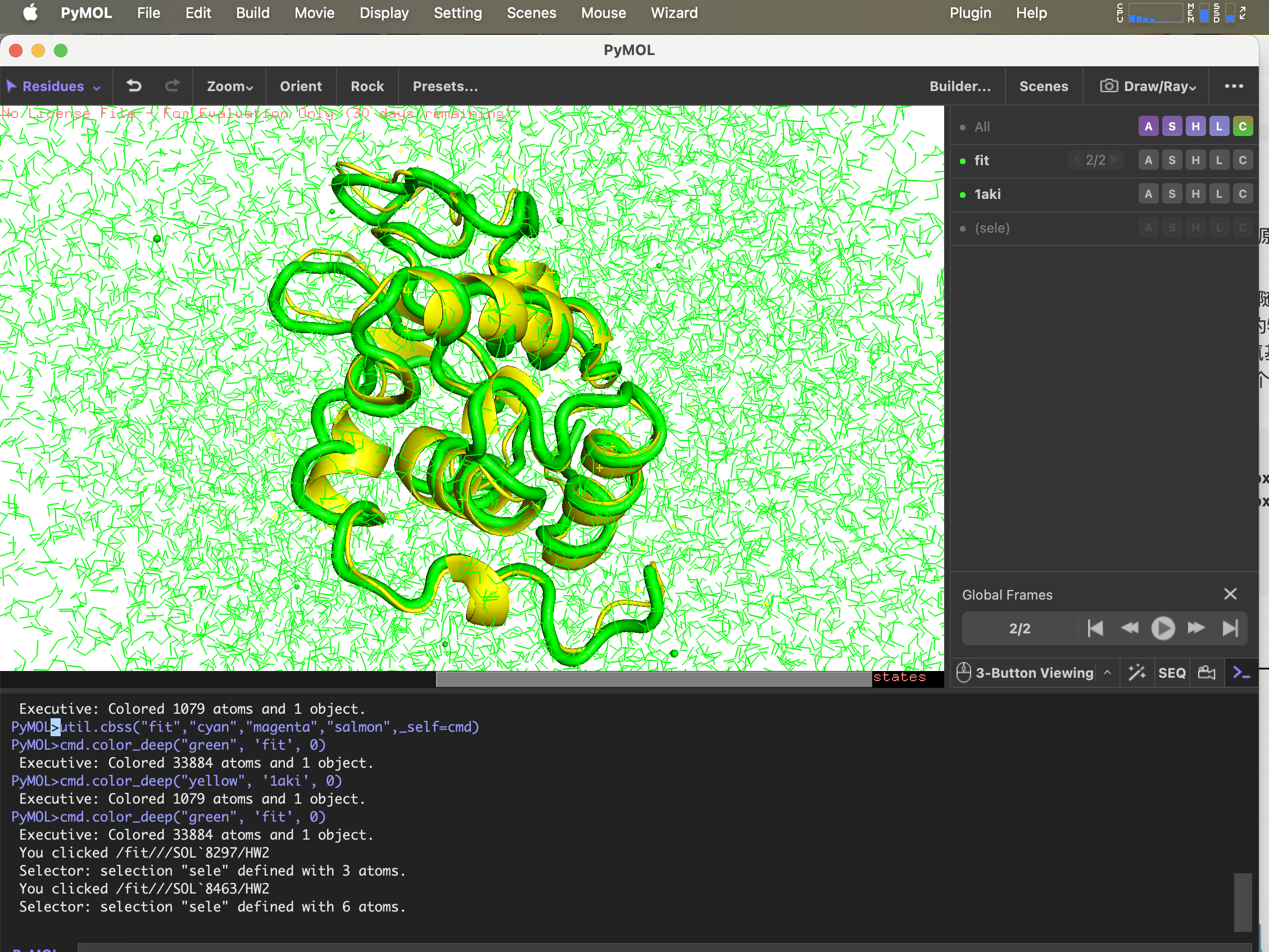
6.g_confrms Comparaison des différences conformationnelles
Pour comparer la structure après simulation avec la structure du fichier PDB d'origine,fit.pdb Un fichier contenant deux structures moléculaires, sélectionnez les deux 4 (Backbone)
gmx_mpi confrms -f1 1AKI_clean.pdb -f2 md_0_1.gro -o fit.pdb
structure pdb ouverte avec pymol
7.g_rmsf Calculez la fluctuation quadratique moyenne (RMSF) et la structure moyenne dans les 500 ps suivantes.g_rmsf Le résultat est une courbe qui change avec le numéro atomique. Sélectionner 1 Protein
Les fluctuations des résidus d’acides aminés peuvent être déduites à l’aide du paramètre RMSF, qui explique l’écart moyen de chaque résidu/atome d’acide aminé par rapport à une position de référence au fil du temps. On peut plutôt dire qu’il analyse des parties spécifiques de la structure d’une protéine qui fluctuent par rapport à sa structure moyenne. Les acides aminés ou les groupes d'acides aminés avec des valeurs RMSF élevées indiquent que le complexe a une plus grande flexibilité, tandis que les acides aminés avec des valeurs RMSF plus faibles indiquent que le complexe a moins de flexibilité. Des fluctuations fréquentes entraînent une moins bonne stabilité. La valeur RMSF est un paramètre dynamique utilisé pour mesurer la flexibilité moyenne de la chaîne principale de chaque position de résidu[19].
gmx_mpi rmsf -s md_0_1.tpr -f md_0_1_noPBC.xtc -b 500 -o fws-rmsf.xvg -ox fws-avg.pdb
gmx_mpi rmsf -s md_0_1.tpr -f md_0_1_noPBC.xtc -b 500 -o fws-rmsf.xvg -ox fws-avg.pdb -res
dit xvg_show -f fws-rmsf.xvg -o rmsf_res.png
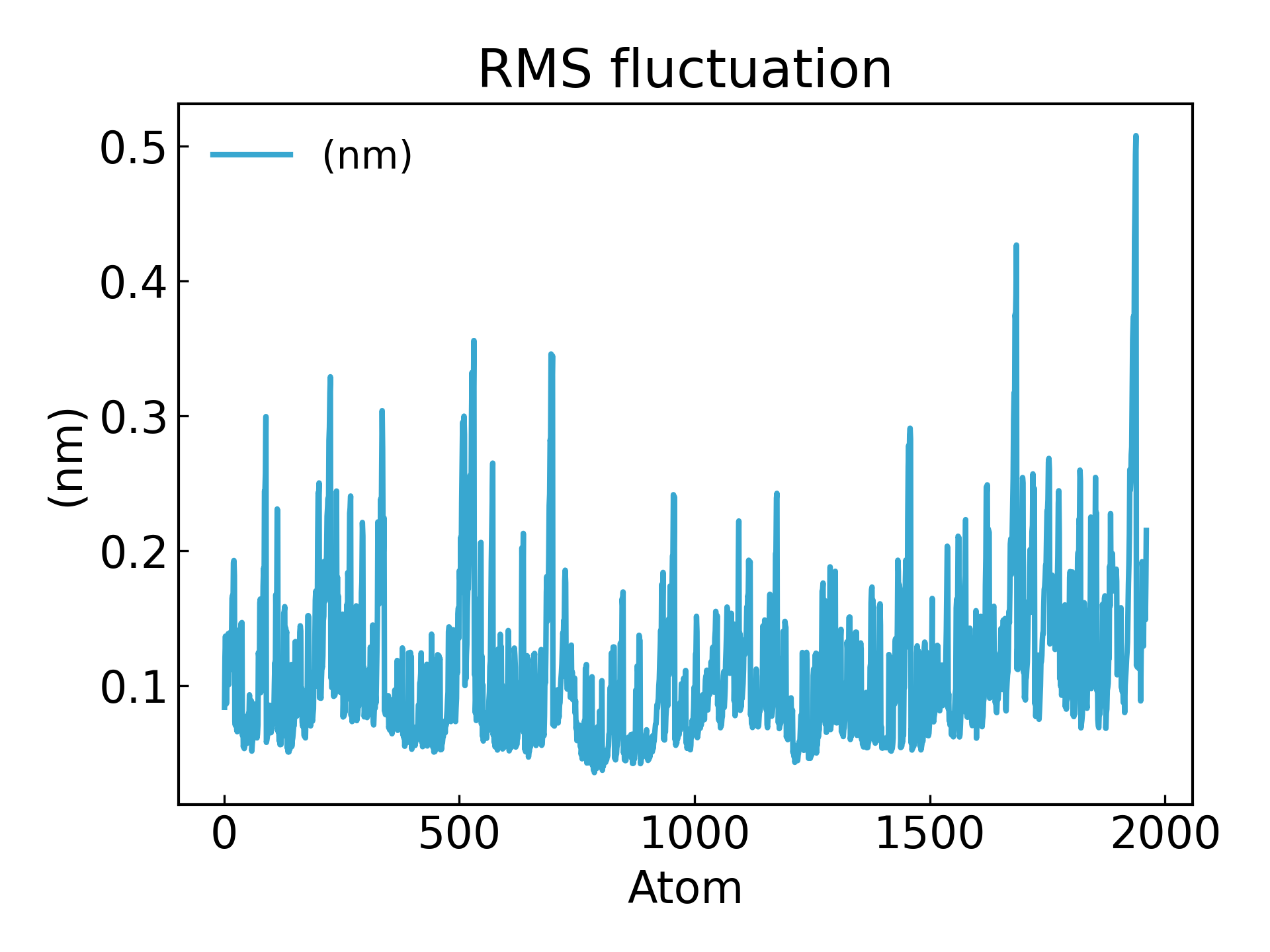
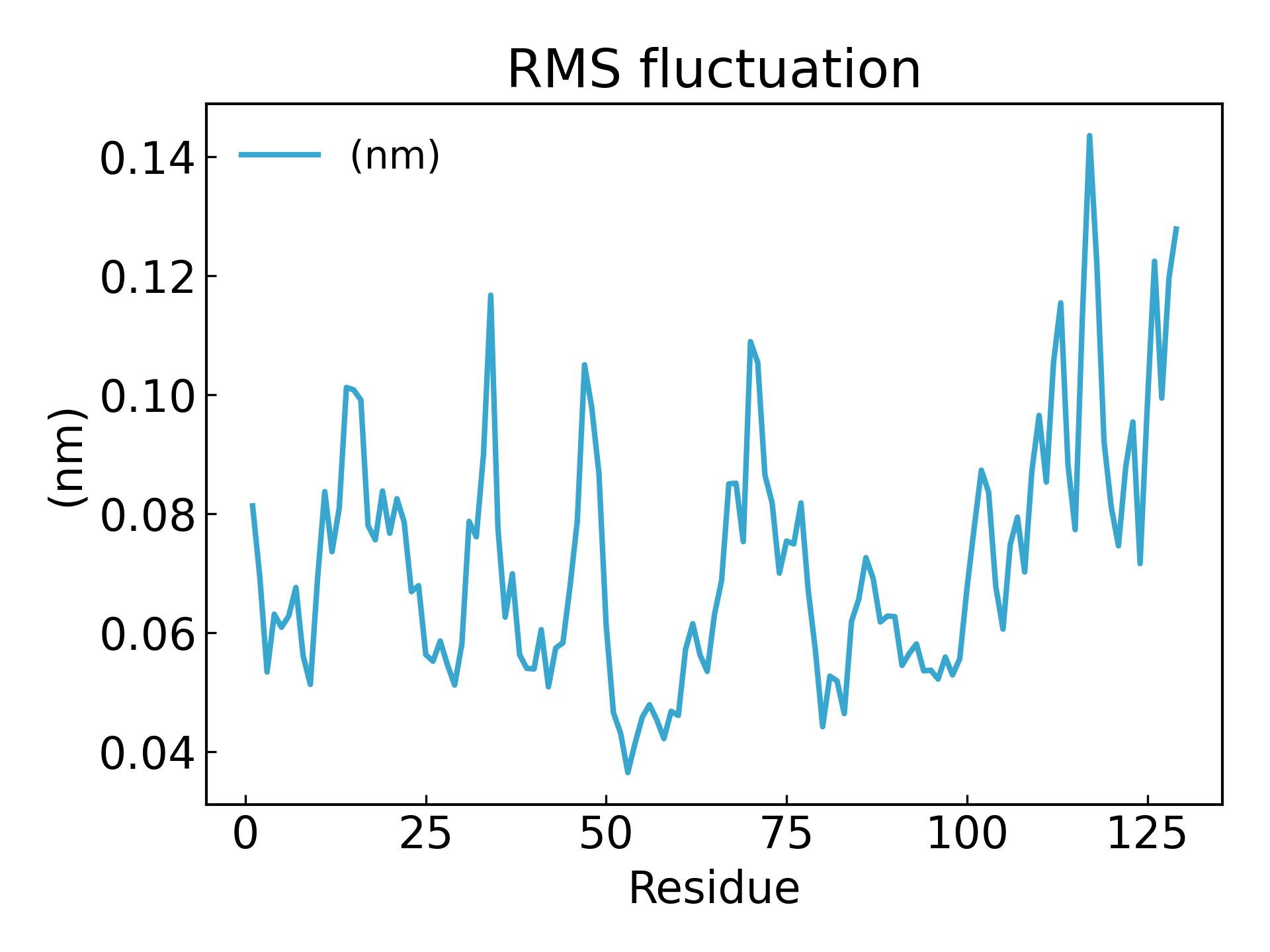
# 最后可以放到 pymol 里面看轨迹动画,点击播放即可
gmx_mpi trjconv -s md_0_1.tpr -f md_0_1_noPBC.xtc -o trajectory.pdb -skip 10
Référence
Créer de l'IA avec l'IA
De l'idée au lancement — accélérez votre développement IA avec le co-codage IA gratuit, un environnement prêt à l'emploi et le meilleur prix pour les GPU.