Command Palette
Search for a command to run...
Tutoriel En Ligne | Les Coûts De Calcul Chutent ! Apple Lance Ml-simplefold, Un Modèle De Repliement Des Protéines Basé Sur La Correspondance De flux.

En septembre 2025, Apple a lancé Ml-simplefold, un modèle d'IA léger de prédiction du repliement des protéines. Premier modèle de repliement des protéines basé sur la correspondance de flux, il a été testé dans des benchmarks de référence tels que CAMEO22 et CASP14.Après avoir réduit le coût de calcul, SimpleFold affiche toujours des performances comparables aux modèles haut de gamme tels que AlphaFold2 et RoseTTAFold2.Dans le même temps, sa version plus petite, SimpleFold-100M, est également compétitive.
Bien que les modèles traditionnels de repliement des protéines offrent une précision impressionnante, ils reposent sur un grand nombre de conceptions architecturales spécifiques à un domaine et de fonctionnalités personnalisées. Les modules de mise à jour triangulaires couramment utilisés, les mécanismes explicites de représentation par paires et les multiples objectifs d'entraînement de ces modèles rendent l'apprentissage coûteux en calcul. De plus, l'architecture et le matériel manquent d'évolutivité, ce qui complique la génération de structures diverses ou la prédiction d'ensemble.
Visant les défauts communs des méthodes traditionnelles,SimpleFold propose un cadre génératif général basé uniquement sur Transformer, brisant la dépendance des modèles de repliement des protéines aux architectures complexes :
* Basé sur la technologie Flow Matching, il ignore les modules complexes tels que l'alignement de séquences multiples (MSA) et génère directement la structure tridimensionnelle des protéines à partir du bruit aléatoire, réduisant ainsi considérablement les coûts de calcul ;
* Adopter une architecture générale qui abandonne des modules spécifiques tels que la mise à jour des triangles et la représentation des paires, en utilisant uniquement le transformateur standard et en améliorant la perception de la structure grâce à des couches adaptatives ;
* Des contraintes structurelles ont été ajoutées pour conférer au modèle une plus grande flexibilité et une cohérence physique accrue lors de la génération de structures tridimensionnelles de protéines. Le modèle à 3 milliards de paramètres, entraîné sur 9 millions de données structurelles, peut fonctionner sans problème sur du matériel grand public, réduisant ainsi considérablement le seuil de calcul.
« Ml-simplefold : un modèle d'IA léger de prédiction du repliement des protéines » est désormais disponible sur le site web HyperAI Hyperneuron (hyper.ai) dans la section « Tutoriels ». Exécutez-le en un clic pour découvrir le dernier outil de génération de protéines.
Lien vers le tutoriel:
Essai de démonstration
- Saisissez l'URL hyper.ai dans votre navigateur. Après être arrivé sur la page d'accueil, cliquez sur la page « Tutoriel », sélectionnez « Modèle de repliement des protéines basé sur la correspondance de flux », puis cliquez sur « Exécuter ce tutoriel en ligne ».
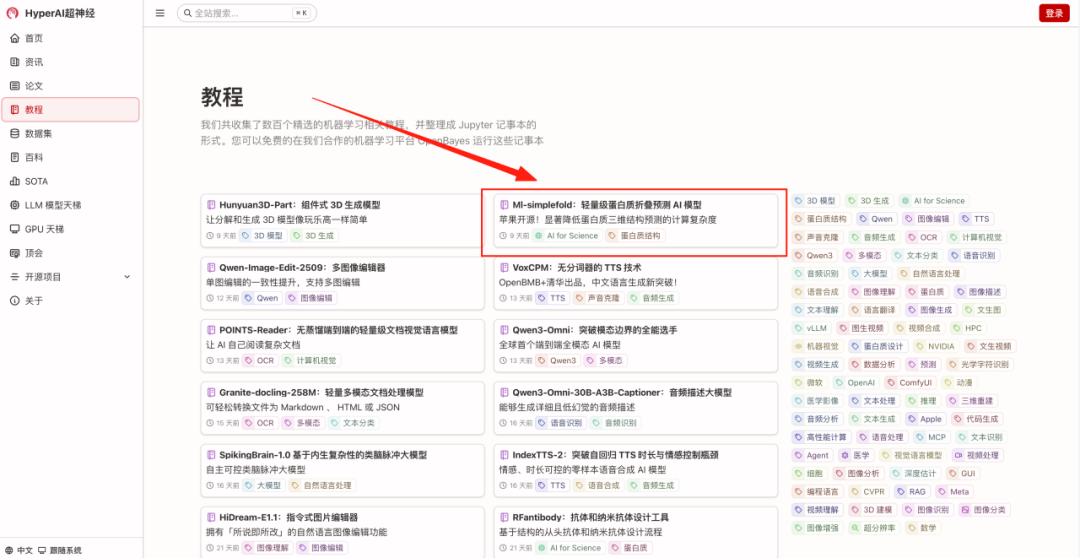
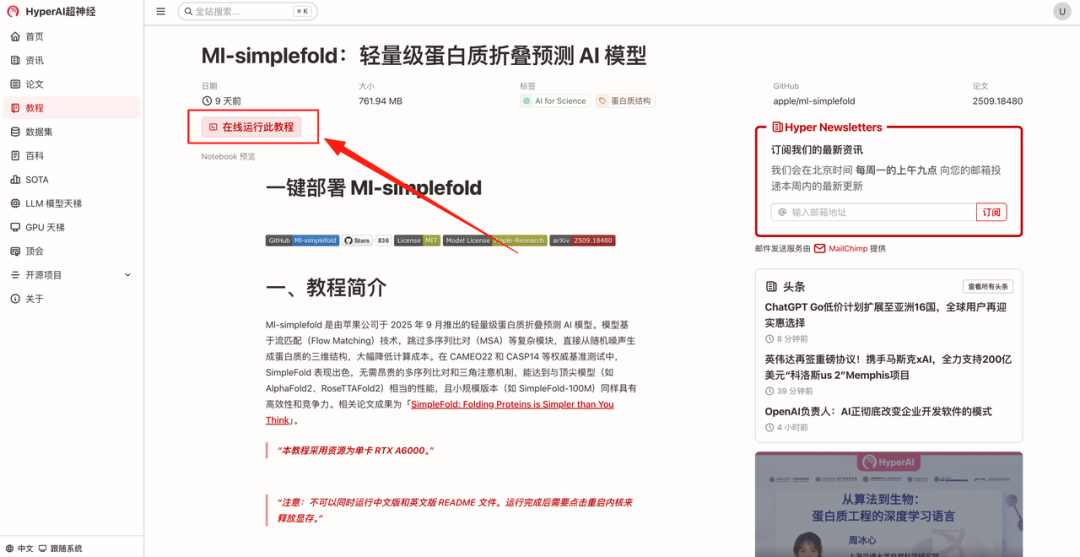
- Une fois la page affichée, cliquez sur « Cloner » dans le coin supérieur droit pour cloner le didacticiel dans votre propre conteneur.
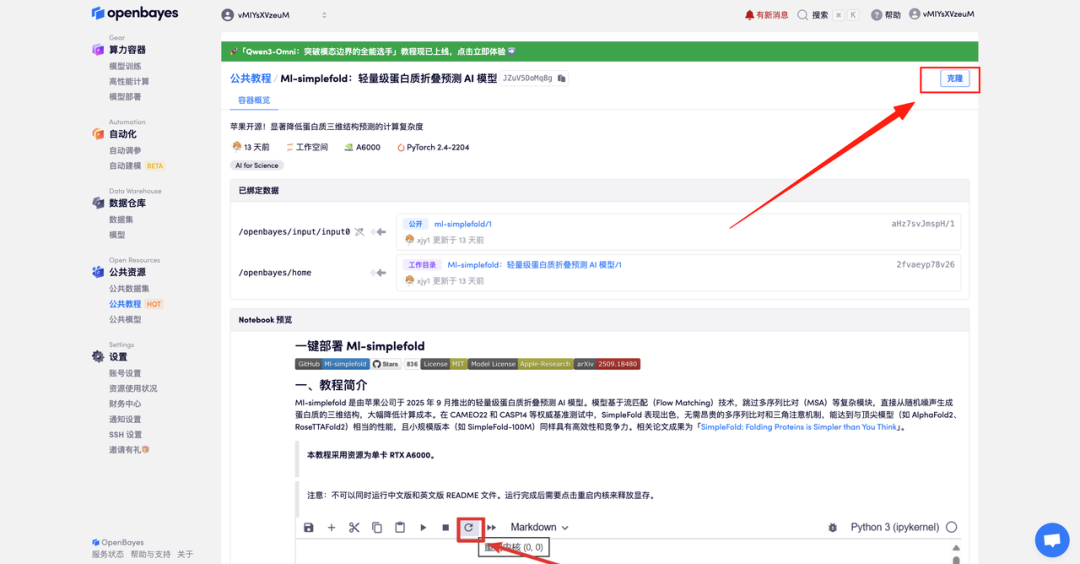
- Sélectionnez les images « NVIDIA RTX A6000 48 Go » et « PyTorch », puis cliquez sur « Continuer ». La plateforme OpenBayes propose quatre options de facturation : paiement à l'utilisation ou forfait journalier, hebdomadaire ou mensuel. Les nouveaux utilisateurs peuvent s'inscrire via le lien d'invitation ci-dessous pour recevoir 4 heures de carte graphique RTX 4090 et 5 heures de temps processeur gratuits !
Lien d'invitation exclusif HyperAI (copier et ouvrir dans le navigateur) :
https://openbayes.com/console/signup?r=Ada0322_NR0n
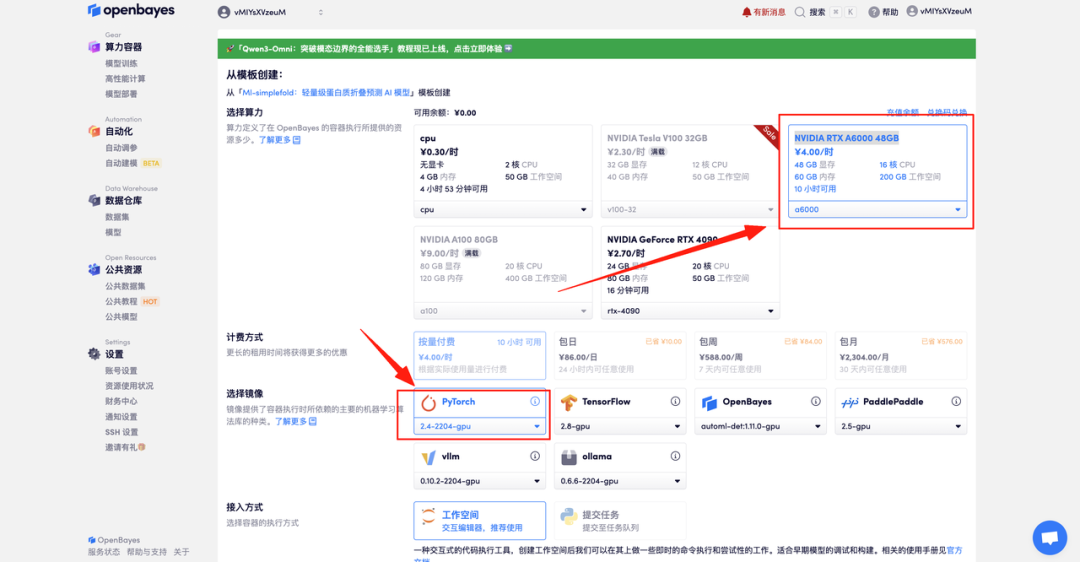
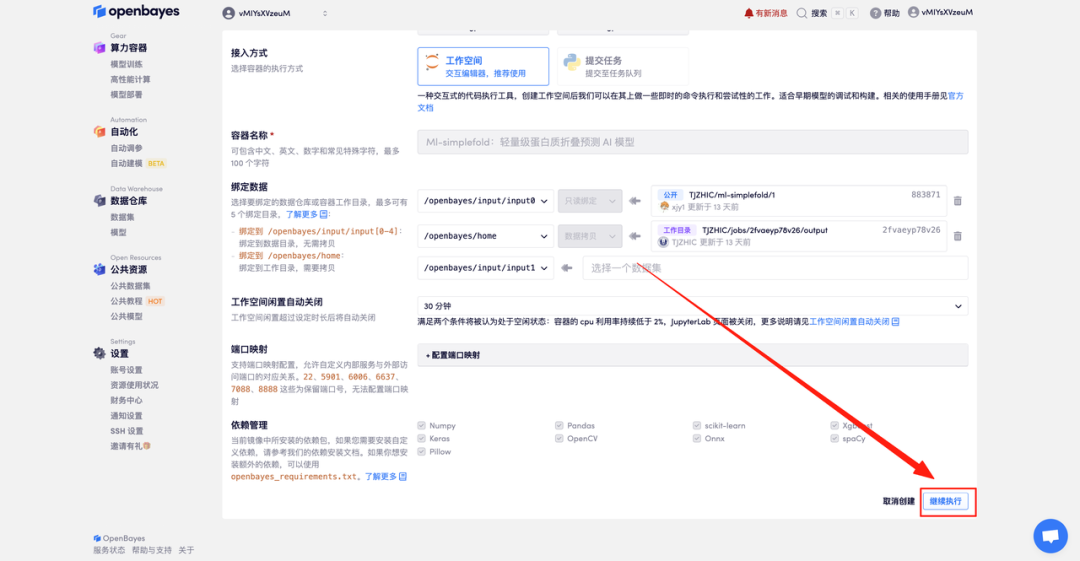
4. Attendez que les ressources soient allouées. Le premier clonage prendra environ 2 minutes. Lorsque le statut passe à « En cours d'exécution », cliquez sur « Ouvrir l'espace de travail » pour accéder à la page de démonstration.
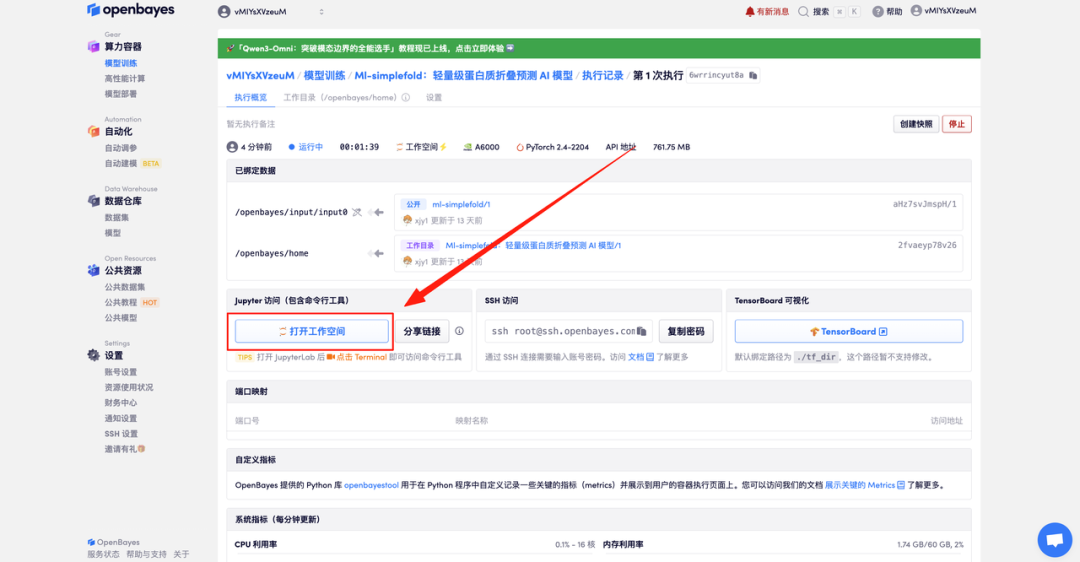
Démonstration d'effet
Voici la page d'utilisation de ML-simplefold. Cliquez sur « README » pour accéder à l'interface de génération.
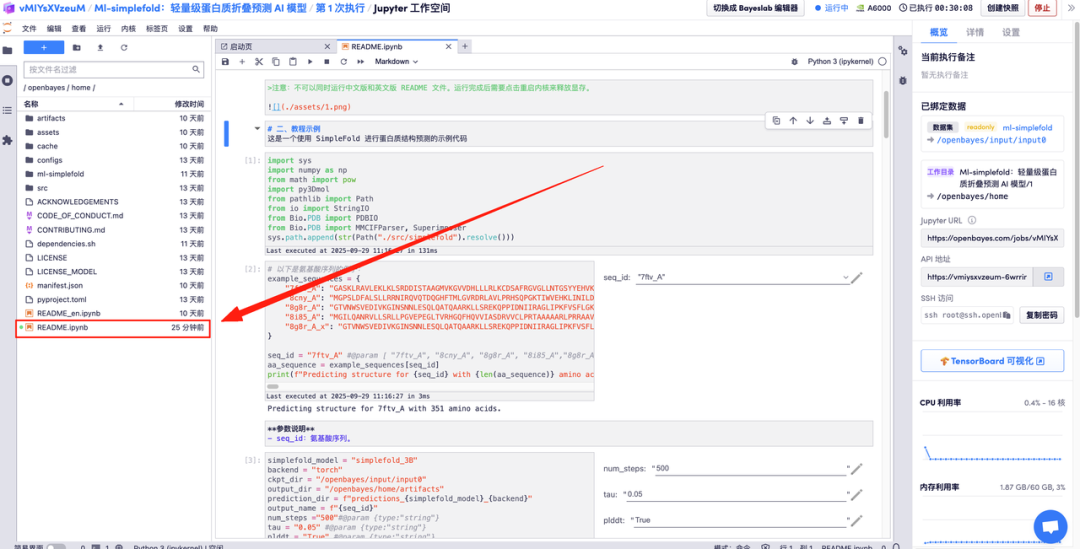
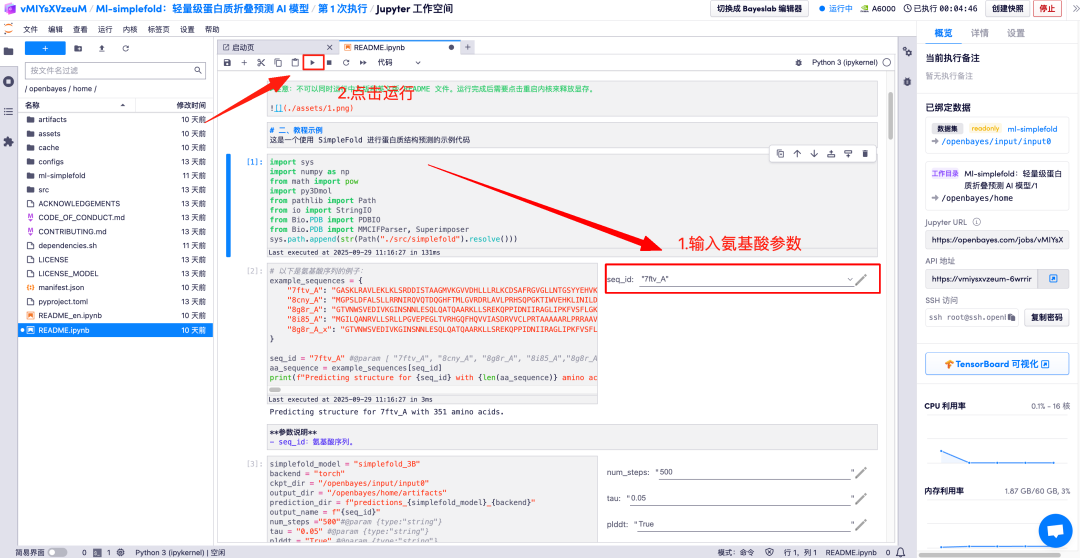
Saisissez les descriptions des paramètres d'acides aminés dans les zones correspondantes et cliquez sur Exécuter pour obtenir le fichier protéique prédit, la structure atomique complète et l'image de visualisation 3D fusionnée avec la structure GT.
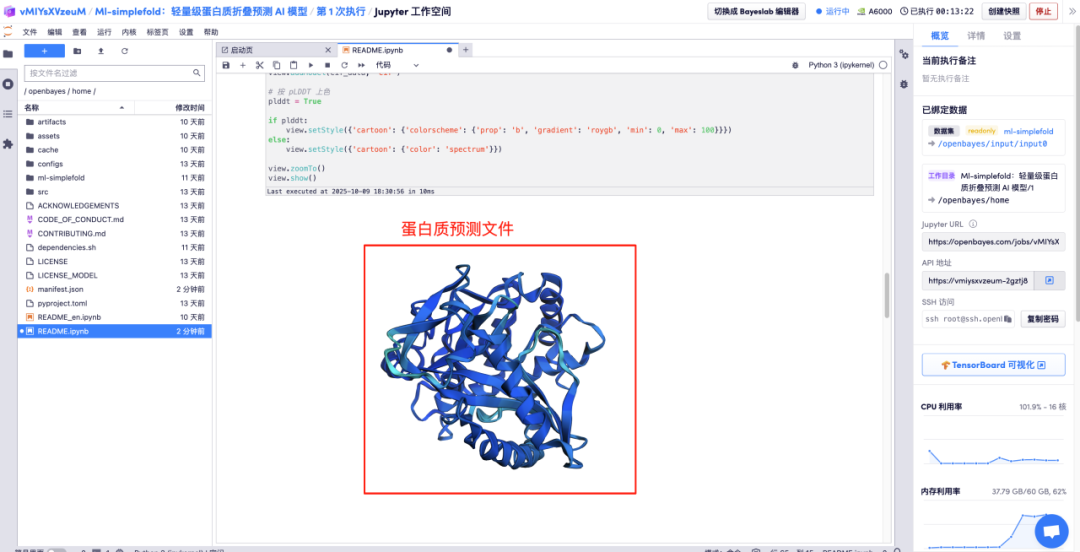
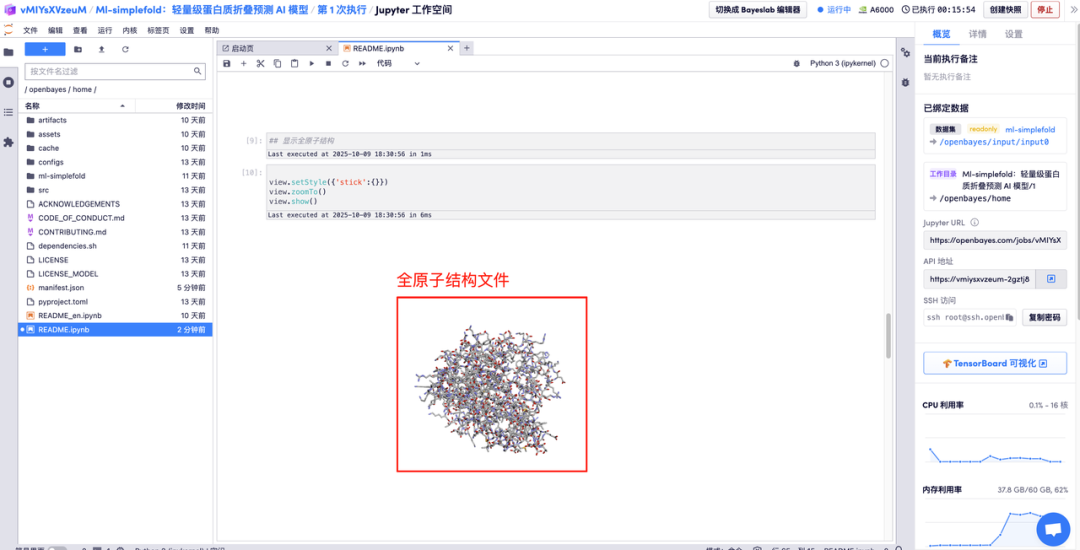
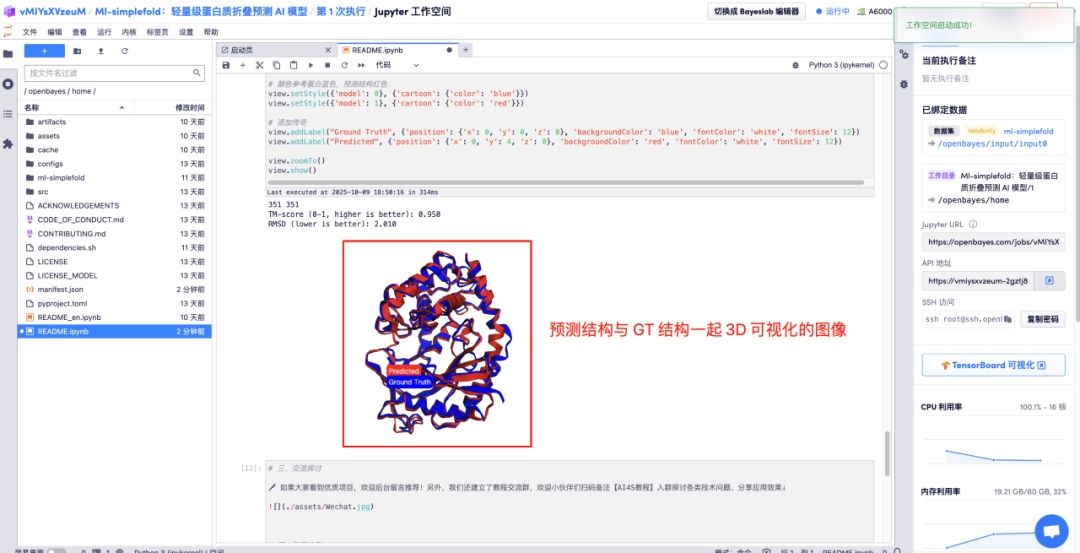
Le tutoriel ci-dessus est celui recommandé par HyperAI cette fois-ci. Bienvenue à tous pour le découvrir !
Lien du tutoriel :








