Command Palette
Search for a command to run...
Du « Criblage À l'aveugle » Au « Positionnement Précis », Le Cadre AlphaPPIMI Améliore Considérablement Les Capacités De Généralisation Et Les Performances De Prédiction Des Régulateurs d'interface PPI Surpassent Les Méthodes existantes.

Au sein des réseaux régulateurs complexes du vivant, les interactions protéine-protéine (IPP) coordonnent la signalisation intracellulaire, le métabolisme énergétique et l'activité des gènes, essentiels au maintien d'une vie normale. Les IPP jouent un rôle central dans le maintien de l'homéostasie physiologique en bonne santé et dans les changements anormaux qui surviennent au cours de la maladie. Des études ont montré que le dysfonctionnement des IPP est étroitement associé au cancer, aux maladies neurodégénératives et à diverses maladies infectieuses. Par conséquent, le développement de médicaments ciblant les IPP est devenu un axe clé de la recherche et du développement de nouveaux médicaments.
Les premiers scientifiques ont étudié les interactions protéine-protéine telles que MDM2-p53 et ont confirmé qu’intervenir dans ces interactions avait le potentiel de traiter des maladies, en fournissant notamment de nouvelles idées pour des cibles de maladies qui étaient auparavant difficiles à cibler.Cependant, la particularité des IPP est que leurs interfaces d’interaction sont généralement relativement plates et manquent de caractéristiques structurelles claires adaptées à l’intégration de médicaments à petites molécules, ce qui pose un énorme défi au développement de médicaments.En particulier pour les IPP nouvellement découverts ou ceux dont les informations structurelles sont limitées, il est plus difficile de trouver des molécules capables de réguler leurs fonctions.
Les chercheurs ont découvert que, bien que l'interface des IPP soit large et plane, il existe encore des zones clés, appelées « points chauds ». Ces zones agissent comme des « interrupteurs » dans l'interaction et constituent des cibles idéales pour la conception de médicaments.
Grâce au développement rapide des technologies d'intelligence artificielle, notamment de l'apprentissage automatique et de l'apprentissage profond, le processus de développement de médicaments pour les IPP s'est considérablement accéléré. Divers algorithmes et outils innovants ont vu le jour, notamment 2P2IHUNTER, qui identifie efficacement les inhibiteurs potentiels des IPP ; PPIMpred, qui permet un criblage virtuel à grande échelle ; et SMMPPI, qui non seulement prédit les molécules régulatrices, mais démontre également une valeur pratique dans la recherche anti-COVID-19. Malgré des progrès significatifs, des défis subsistent. Les méthodes informatiques traditionnelles, qui s'appuient fortement sur le criblage par similarité, peinent à saisir pleinement les caractéristiques d'interaction complexes des interfaces des IPP. De plus, les modèles existants ont des capacités de généralisation limitées entre différents types de protéines, ce qui entrave l'efficacité du développement de médicaments pour de nouvelles cibles.
Ces dernières années, les modèles de langage pré-entraînés basés sur Transformer ont apporté de nouvelles solutions aux problèmes mentionnés ci-dessus. Ces modèles peuvent apprendre automatiquement des caractéristiques clés d'un grand nombre de séquences protéiques, prédisant ainsi les interactions plus intelligemment.
Sur la base de cette orientation, l'équipe de recherche conjointe de l'Université chinoise du pétrole et de l'Université Yonsei a intégré plusieurs technologies avancées pour construire un nouveau cadre appelé AlphaPPIMI.Cet outil combine des modèles pré-entraînés à grande échelle et des mécanismes d'apprentissage adaptatif pour relever le défi principal de « découvrir des régulateurs qui ciblent spécifiquement l'interface des PPI ».En exploitant pleinement les avantages des modèles à grande échelle pré-entraînés et en modélisant efficacement des modèles de liaison complexes grâce à un module d'attention croisée dédié, la capacité de généralisation du modèle sur différentes familles d'IPP a été considérablement améliorée, offrant un soutien solide au développement futur de médicaments ciblant les IPP.
Les résultats de recherche pertinents ont été publiés dans le Journal of Cheminformatics sous le titre « Alphappimi : un cadre complet d'apprentissage en profondeur pour prédire les interactions PPI-modulateur ».

Adresse du document :
https://jcheminf.biomedcentral.com/articles/10.1186/s13321-025-01077-2
Suivez le compte officiel et répondez « AlphaPPIMI » pour obtenir le PDF complet
Autres articles sur les frontières de l'IA :
https://hyper.ai/papers
Ensemble de données : Construction d'un système d'ensemble de données PPI avec DLiP comme noyau
L’étude a utilisé l’ensemble de données DLiP comme noyau de formation, qui contient 120 PPI et leurs 12 605 régulateurs uniques correspondants.Il peut également fournir la séquence, la structure tridimensionnelle et les données d'activité expérimentale de chaque paire de complexes protéiques, offrant ainsi un support complet pour la construction de modèles.
Afin de réaliser une validation indépendante, l'équipe de recherche a construit deux ensembles de tests de référence à partir des bases de données DiPPI et iPPIDB. Ces deux ensembles contiennent des modulateurs d'interface validés expérimentalement, ainsi que leurs structures et informations de liaison. Lors de la compilation des données, l'équipe a mis en œuvre trois contrôles qualité : conserver uniquement les IPP hétérodimériques, éliminer les échantillons dont les sites de liaison sont incertains et limiter le champ d'application aux IPP humains. De plus, les composés agissant sur plusieurs cibles ont été séparés afin de garantir une annotation précise.
Les détails finaux des deux ensembles de référence sont les suivants :DiPPI contient 201 régulateurs correspondant à 1 316 cibles PPI.Chaque échantillon possède une structure moléculaire, une séquence protéique, une structure d'interface et une étiquette active ; iPPIDB couvre 2 203 régulateurs et 34 IPP, et toutes les séquences protéiques proviennent de la base de données UniProt, garantissant la cohérence des données.
Après avoir analysé les propriétés physiques et chimiques des deux ensembles de référence, il a été constaté qu'ils différaient considérablement en termes de caractéristiques de ciblage d'interface et de distribution de l'espace chimique, ce qui augmenterait la difficulté de généralisation du modèle.En calculant l’empreinte moléculaire ECFP4, nous avons également constaté que la similarité Tanimoto moyenne des composés dans les deux ensembles de référence est très faible.Cela indique que la diversité structurelle de ces composés est relativement élevée.
Pour une famille d'IPP donnée, l'étude a également sélectionné des régulateurs sélectifs pour d'autres familles d'IPP comme échantillons potentiellement inactifs, tout en excluant les molécules ayant des structures similaires à celles des régulateurs actifs connus afin de réduire le risque de faux négatifs. Compte tenu du déséquilibre entre le nombre d'échantillons positifs et négatifs, l'équipe a sous-échantillonné les échantillons négatifs afin de constituer un ensemble de données équilibré. Une analyse de sensibilité ultérieure a montré que, quel que soit l'ajustement du ratio d'échantillons positifs et négatifs, les performances du modèle étaient très stables et peu dépendantes de ce ratio. Il convient de noter que, bien que des composés inactifs aient été vérifiés, ils n'ont pas été inclus dans l'ensemble d'échantillons négatifs car leur répartition est inégale et peut entraîner un biais dans les données après leur inclusion.
Afin de vérifier la valeur d'application pratique de cette méthode,L'équipe de recherche a également examiné la « bibliothèque spécifique aux IPP » dans la base de données ChemDiv - cette bibliothèque contient 205 497 composés spécifiquement conçus pour les propriétés interfaciales des IPP.Ce criblage virtuel à grande échelle a démontré la praticité de cette méthode dans les scénarios de développement de médicaments.
Cadre AlphaPPIMI : extraction de fonctionnalités multi-sources, attention croisée bidirectionnelle et optimisation de la généralisation CDAN
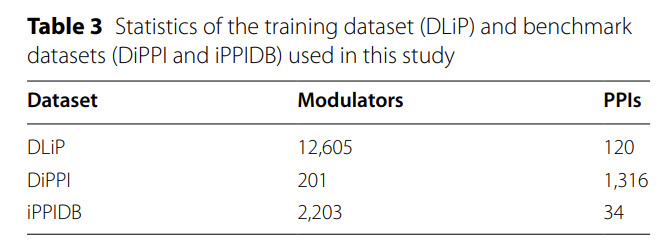
Comme le montre la figure ci-dessous,Cette étude a développé un nouveau cadre informatique, AlphaPPIMI, spécifiquement pour prédire la relation de liaison entre les IPP et les régulateurs.En mettant l'accent sur le ciblage des interactions au niveau des sites de liaison d'interface, ce cadre intègre plusieurs modules avancés, notamment Uni-Mol2, ESM2, ProTrans, ECFP et PFeature, s'efforçant d'extraire de manière exhaustive les fonctionnalités liées au PPI tout en réalisant un apprentissage de représentation efficace.
Dans la phase de caractérisation moléculaire, l’équipe de recherche a utilisé le modèle Uni-Mol2 avec 84 millions de paramètres pour intégrer les atomes, les liaisons chimiques, les informations géométriques et les empreintes moléculaires.Un vecteur de caractéristiques globales de 768 dimensions a été généré pour chaque modulateur. L'équipe a également intégré les empreintes digitales ECFP4 pour générer un vecteur binaire de 1 024 dimensions afin de capturer des informations chimiques clés, telles que la sous-structure cyclique. Finalement, ces deux types de caractéristiques ont été combinés pour produire un vecteur de caractéristiques de 1 792 dimensions englobant la topologie moléculaire, la géométrie 3D et la sous-structure chimique, fournissant des données fiables pour les prédictions de liaison d'interface.
L'extraction des caractéristiques des protéines utilise trois approches complémentaires :Le modèle ESM2-150M, basé sur l'architecture Transformer, a été entraîné sur 60 millions de séquences UniRef50 et génère des vecteurs de caractéristiques à 640 dimensions capturant spécifiquement les relations entre les acides aminés liées à la formation des interfaces. Le modèle ProtTrans, entraîné sur plus de 45 millions de séquences protéiques, produit des vecteurs d'inclusion à 1 024 dimensions capturant des schémas évolutifs complémentaires à ESM2. Enfin, la méthode PFeature fournit des informations sur la structure et les propriétés physicochimiques des protéines grâce à 19 catégories de descripteurs. La fusion de ces trois méthodes génère une représentation protéique à 3 366 dimensions qui couvre de manière exhaustive les schémas de séquences protéiques et les propriétés spécifiques aux interfaces.
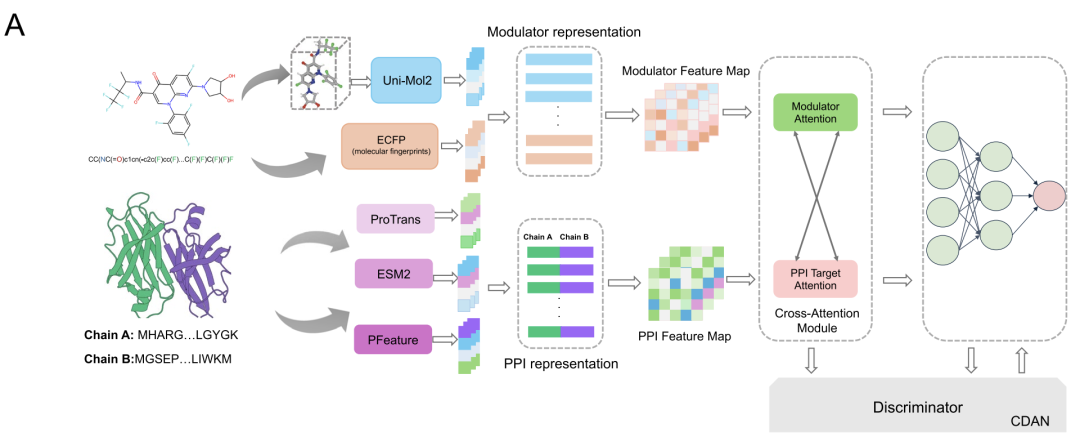
Pour modéliser les interactions complexes entre protéines et régulateurs, AlphaPPIMI a conçu un module d'attention croisée bidirectionnel, illustré dans la figure ci-dessous. Ce module effectue d'abord des transformations linéaires sur la matrice de caractéristiques du régulateur FM et la matrice de caractéristiques de la cible FP, qui sont ensuite intégrées au sous-module d'attention, permettant ainsi un échange bidirectionnel d'informations au niveau clé-valeur. Les caractéristiques PPI sont optimisées à l'aide des pondérations d'attention de la source du régulateur, tandis que les caractéristiques du régulateur sont ajustées grâce au mécanisme d'attention piloté par PPI.Des connexions résiduelles et des opérations de pooling maximales sont également ajoutées au module.Il peut apprendre de manière dynamique le modèle d'interaction entre les deux tout en conservant les informations uniques de chaque modalité et, en fin de compte, produire une représentation plus complète de l'interaction.
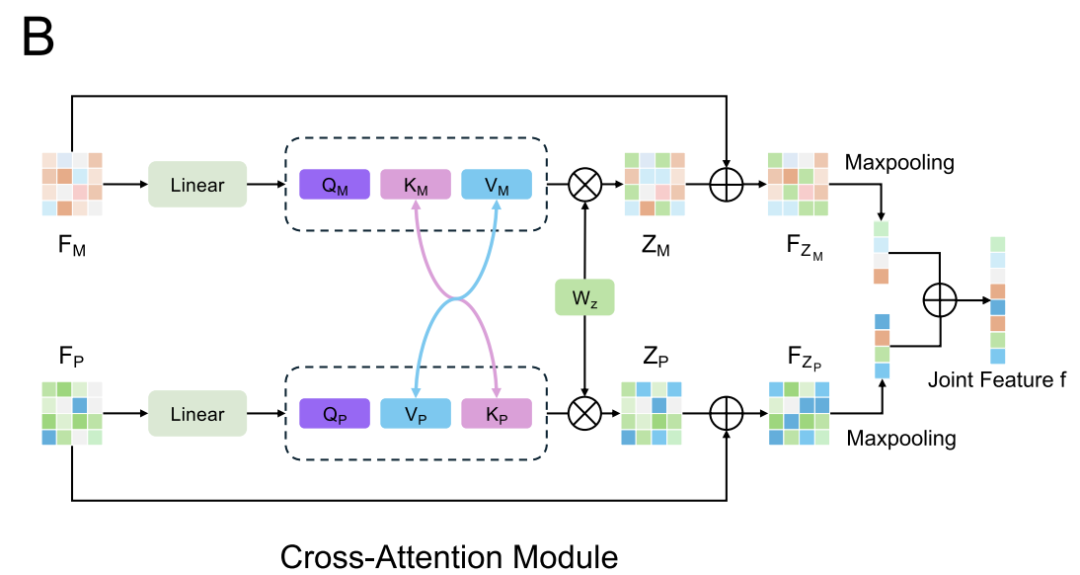
Compte tenu des différences de distribution des caractéristiques entre les jeux de données (par exemple, DiPPI se concentre sur les modulateurs ciblés par interface, tandis que les jeux de données généraux comme DLiP manquent de telles informations), AlphaPPIMI introduit également un réseau antagoniste conditionnel de domaine (CDAN), comme illustré dans la figure ci-dessous. CDAN utilise une « représentation conjointe de l'intégration des caractéristiques et de la prédiction de classification » comme condition du discriminateur de domaine, préservant ainsi les caractéristiques discriminantes tout en assurant l'alignement de la distribution entre les domaines source et cible.Le processus de formation adopte un jeu minimax : l'encodeur de caractéristiques et le module d'attention croisée sont responsables de la génération de représentations invariantes de domaine, tandis que le discriminateur est utilisé pour distinguer les sources de caractéristiques.Ce mécanisme améliore considérablement la capacité de généralisation du modèle à travers différentes familles de protéines et fournit un support plus robuste pour l’identification de nouveaux régulateurs ciblés sur l’interface.
Évaluation et vérification de l'application de la capacité de généralisation inter-domaines d'AlphaPPIMI
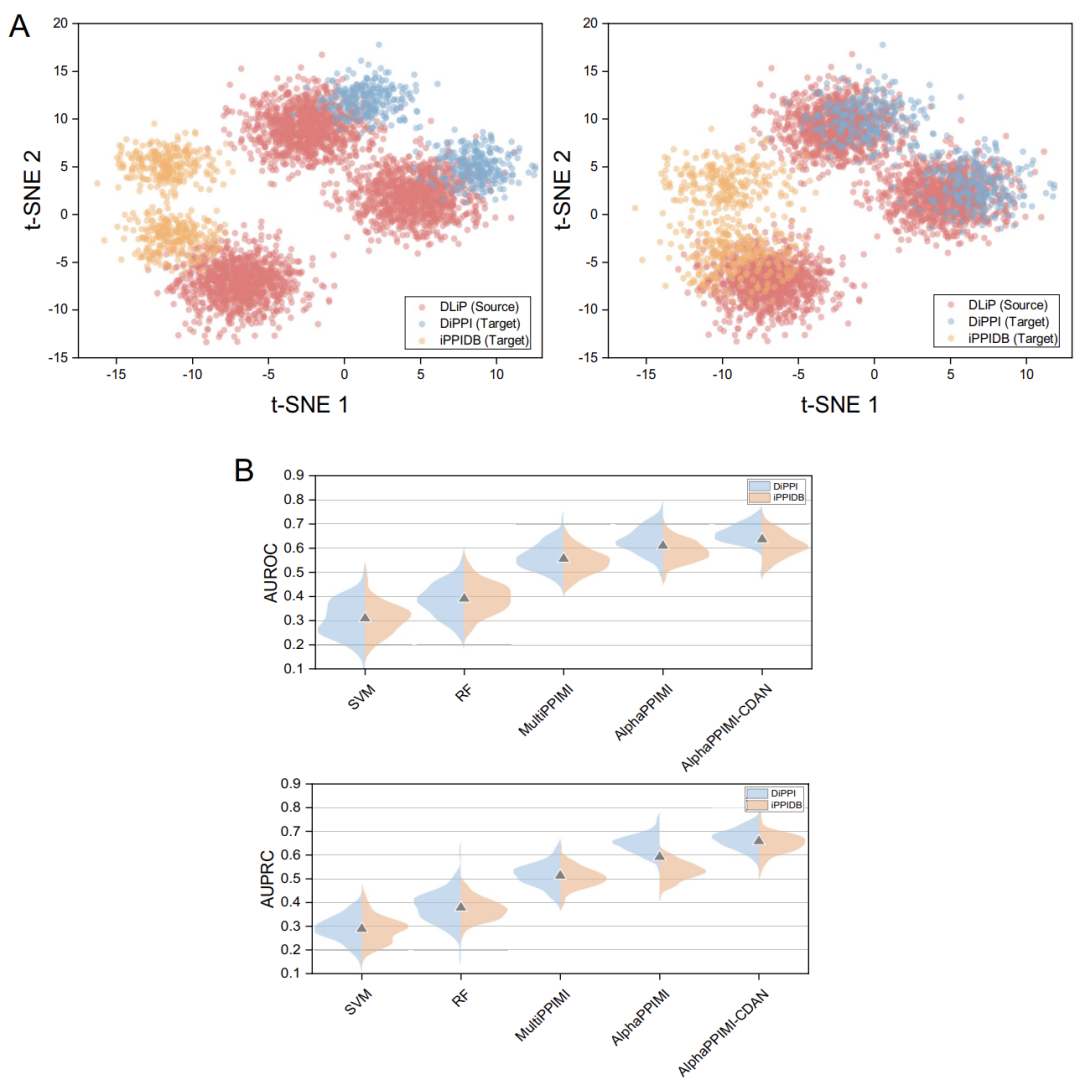
Afin de tester l'adaptabilité inter-domaines d'AlphaPPIMI dans la prédiction des régulateurs des IPP, l'équipe de recherche a conçu une expérience de migration.L'ensemble de données DLiP est considéré comme le « domaine source » (données utilisées pour la formation du modèle), et les ensembles de données DiPPI et iPPIDB sont considérés comme les « domaines cibles » (données utilisées pour la validation du modèle).
Les résultats expérimentaux montrent que les performances de tous les modèles se dégradent en cas de changement de domaine, mais AlphaPPIMI est plus robuste.Ses valeurs AUROC et AUPRC sur DiPPI sont significativement supérieures à celles de MultiPPIMI. L'écart entre les performances inter-domaines et intra-domaines souligne la nécessité de stratégies d'adaptation de domaine. Comme le montre la figure ci-dessous, l'étude a également proposé l'architecture AlphaPPIMI-CDAN, qui permet une adaptation de la distribution inter-domaines grâce à l'alignement conditionnel des caractéristiques. Ce modèle surpasse largement les modèles de base sur DiPPI et iPPIDB. Contrairement aux méthodes traditionnelles d'alignement des bords, cette méthode guide l'alignement des caractéristiques en fonction de la distribution conditionnelle des catégories, générant ainsi des représentations plus discriminantes. Elle permet de traiter efficacement le décalage de distribution causé par de subtiles différences fonctionnelles dans les domaines PPI, tout en atténuant le transfert négatif afin d'améliorer la robustesse et la généralisation des prédictions inter-domaines.
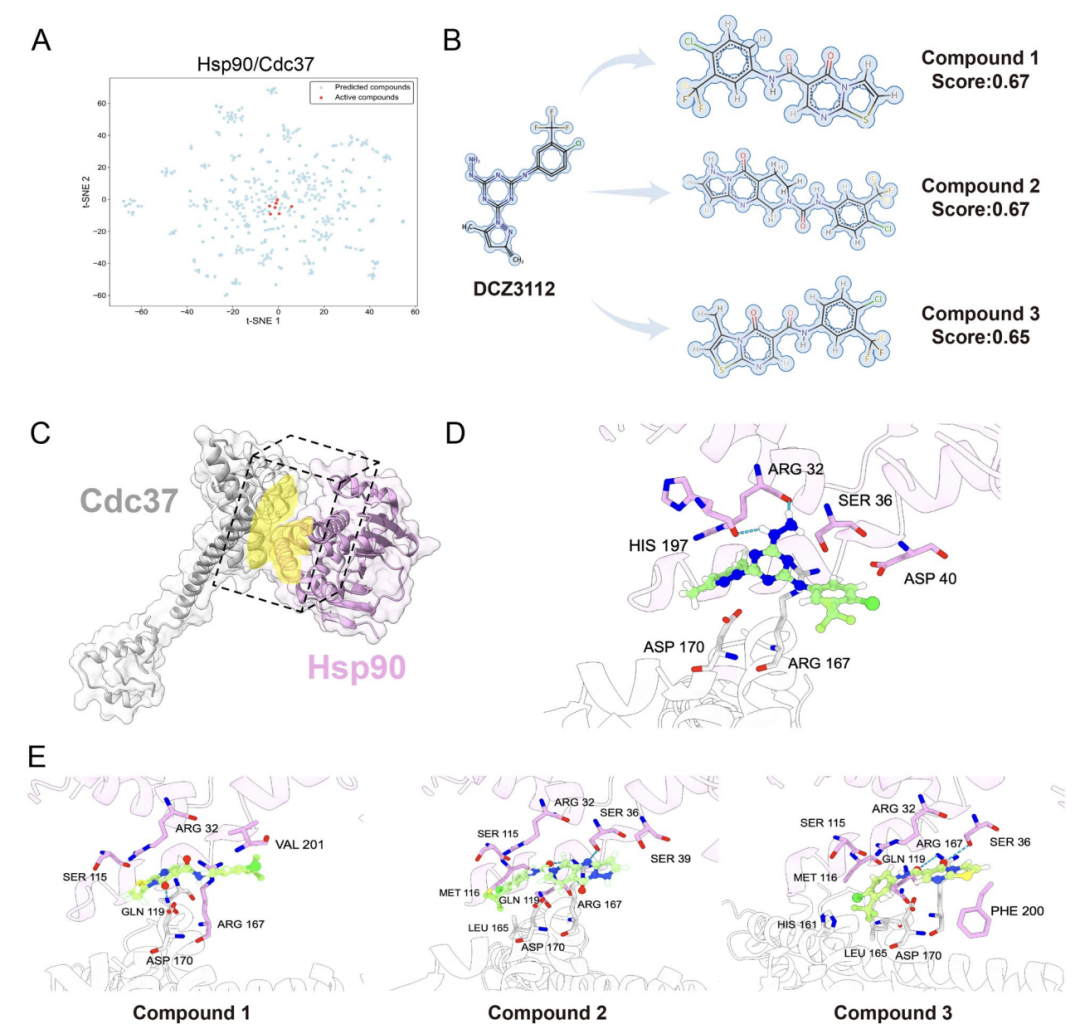
Dans la vérification de l'application pratique, l'étude a également pris comme objet les IPP Hsp90-Cdc37, qui ont une structure d'interface claire et constituent une cible anticancéreuse clé.Comme le montre la figure A ci-dessous, AlphaPPIMI a examiné des composés avec des scores de prédiction > 0,8 dans la bibliothèque ChemDiv, dont l'espace chimique était proche de la distribution des inhibiteurs actifs connus ; comme le montre la figure B ci-dessous, les chercheurs ont utilisé l'inhibiteur vérifié DCZ3112 comme référence et ont examiné trois composés candidats par similarité structurelle et analyse pharmacophore ; comme le montrent les figures D-E ci-dessous, l'amarrage moléculaire a montré que ces composés peuvent former des interactions similaires aux molécules de référence avec des résidus clés tels que Arg32 et Ser36, améliorant ainsi leur potentiel inhibiteur.
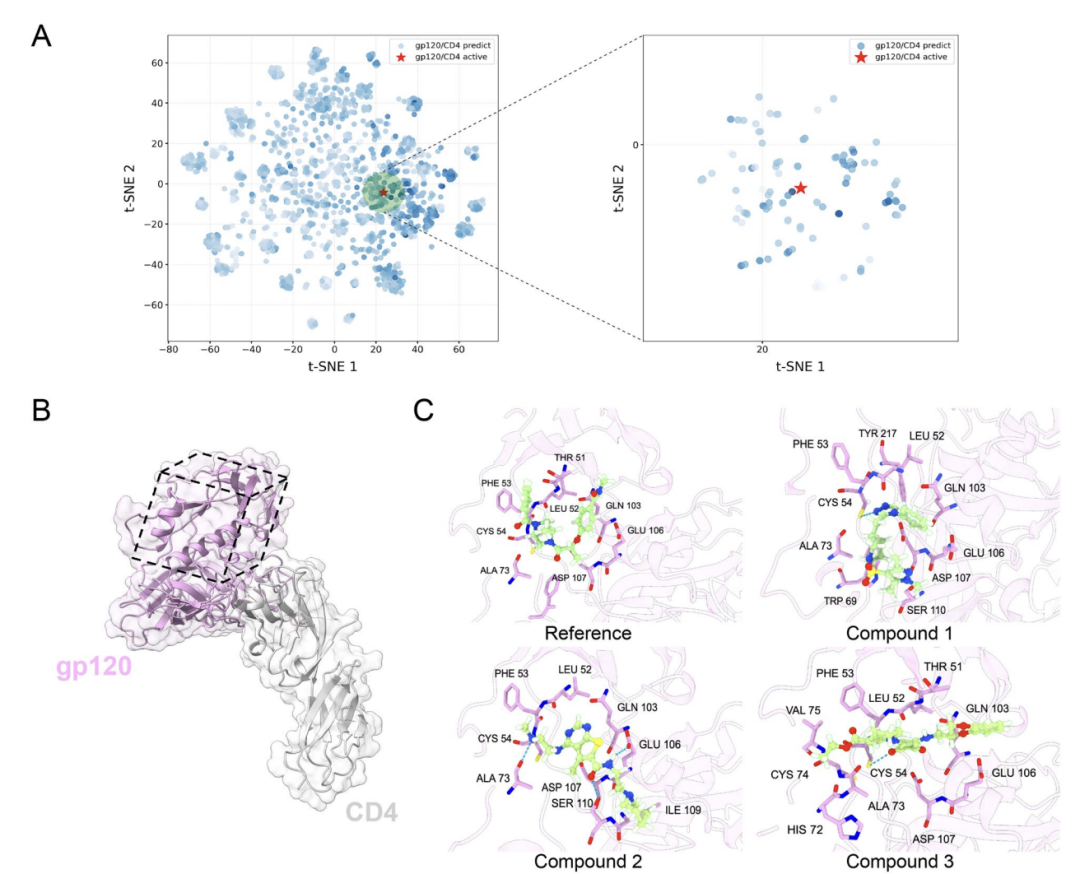
Concernant l'application d'AlphaPPIMI au criblage de modulateurs allostériques des IPP, comme le montre la figure A ci-dessous, les chercheurs ont pris comme exemple l'interaction entre la gp120 du VIH-1 et le CD4. AlphaPPIMI a sélectionné des composés avec une probabilité prédite supérieure à 0,8, dont l'espace chimique chevauchait fortement celui d'inhibiteurs actifs connus. Comme le montre la figure BC ci-dessous, les résultats d'amarrage moléculaire basés sur la structure d'interface atypique (PDB : 6L1Y) ont montré que les composés candidats pouvaient interagir avec des résidus clés tels que THR51, LEU52 et PHE53, indiquant qu'AlphaPPIMI peut découvrir des inhibiteurs allostériques ciblant l'interface d'IPP difficiles à traiter, offrant ainsi une nouvelle approche pour le développement de médicaments apparentés.
La collaboration industrie-université-recherche favorise la diffusion des médicaments ciblés par les IPP, de la recherche fondamentale à l'application clinique
Dans la recherche et le développement de médicaments ciblant les IPP, le monde universitaire et l’industrie travaillent en étroite collaboration pour faire progresser progressivement la recherche fondamentale dans ce domaine vers une application clinique.
À la frontière académique, de nombreuses équipes de recherche explorent des méthodes de prédiction et de ciblage des IPP plus précises et plus efficaces.Par exemple, une équipe de l’Université de Stanford a développé un agent d’IA biomédical général appelé Biomni.Cet agent intelligent est capable d'effectuer de manière autonome des tâches de recherche complexes dans de multiples disciplines biomédicales, notamment la génétique, la génomique, la microbiologie, la pharmacologie et la médecine clinique. La création de Biomni marque la transition de l'IA en recherche biomédicale, passant d'un simple utilisateur d'outils à un décideur autonome. En intégrant des ressources de recherche scientifique dispersées dans des unités comportementales intelligentes et exploitables, Biomni permet non seulement de surmonter les goulots d'étranglement fragmentés des processus de recherche traditionnels, mais favorise également potentiellement l'émergence d'un moteur de découverte scientifique interdisciplinaire, à haut débit et autonome.
Une autre étude représentativeL'Université Sun Yat-sen a proposé une méthode de prédiction des PPI basée sur l'extraction de caractéristiques de fusion et un nouveau mécanisme de sélection de caractéristiques non supervisée.Des expériences approfondies ont démontré que la méthode proposée fonctionnait bien sur cinq ensembles de données couvrant les interactions intra- et inter-espèces, surpassant largement les 16 méthodes d'apprentissage automatique existantes. Cette recherche fournit non seulement un cadre efficace et fiable pour les tâches de prédiction d'IPP à grande échelle, mais démontre également une grande adaptabilité fonctionnelle, offrant une nouvelle solution pour la prédiction des interactions médicament-médicament et médicament-aliment.
En matière de transposition industrielle, les entreprises s'emploient également à faire progresser activement ces avancées scientifiques vers la clinique. Par exemple, l'AN2025 (nom générique : buparlisib), développé par la société biopharmaceutique chinoise Adlai Nortye grâce à une licence mondiale de Novartis, est un pan-inhibiteur ciblant spécifiquement la voie de signalisation PI3K. Il est désormais entré dans un essai clinique mondial de phase III, principalement pour le traitement des patients atteints d'un carcinome épidermoïde de la tête et du cou récidivant ou métastatique ayant progressé malgré un traitement anti-PD-1/PD-L1.
Un autre exemple est Iqirvo (elafibranor), lancé par le célèbre laboratoire pharmaceutique français Ipsen. Premier nouveau traitement contre la CBP approuvé au cours de la dernière décennie, il a confirmé l'intérêt clinique de la régulation des IPP dans le domaine non tumoral et a ouvert la voie à un nouveau paradigme thérapeutique pour les maladies métaboliques complexes. Son approbation a également favorisé une exploration approfondie du réseau d'interactions protéiques de la famille des PPAR.
La collaboration étroite entre le monde universitaire et l'industrie dans le domaine des médicaments ciblant les IPP a non seulement accéléré la traduction des résultats de la recherche scientifique en valeur clinique, mais a également considérablement amélioré l'efficacité et le taux de réussite du développement de nouveaux médicaments. Des modèles prédictifs d'IA multimodaux aux candidats-médicaments aux bénéfices cliniques évidents, cette collaboration interdisciplinaire redéfinit la voie de l'innovation biomédicale. À l'avenir, grâce à l'intégration de davantage de données et d'algorithmes, et au renforcement de la collaboration interinstitutionnelle et interdisciplinaire, les médicaments ciblant les IPP pourraient apporter encore plus de progrès dans le traitement de maladies complexes.
Liens de référence :
1.https://mp.weixin.qq.com/s/ryYJ6T7qEjnjvkhBL4-dAA
2.https://mp.weixin.qq.com/s/7upIPYam1LR0TiGBYXmkOw
3.https://mp.weixin.qq.com/s/69GU1R5lXHdTLttlT8apyw








