Command Palette
Search for a command to run...
Redéfinition De La Classification Des Modèles De Langage Protéique En Fonction De La Relation structure/séquence/fonction : Le Dr Li Mingchen Explique En Détail Les Modèles De Langage protéique.

La troisième « École d'été sur l'IA pour la bio-ingénierie » de l'Université Jiao Tong de Shanghai a officiellement ouvert ses portes du 8 au 10 août 2025.Cette école d'été a réuni plus de 200 jeunes talents, chercheurs scientifiques et représentants de l'industrie de plus de 70 universités, plus de 10 institutions de recherche scientifique et plus de 10 entreprises leaders du secteur à travers le monde, en se concentrant sur le développement intégré de l'intelligence artificielle (IA) et de la bio-ingénierie.
Parmi eux, dans la section du cours « Frontières des algorithmes d'IA », Li Mingchen, chercheur postdoctoral au sein du groupe de recherche Hong Liang de l'Institut des sciences naturelles de l'Université Jiao Tong de Shanghai, a partagé avec tous les résultats de pointe des modèles de langage protéique dans la prédiction de fonction, la génération de séquence, la prédiction de structure, etc., avec le thème « Modèle de base des protéines et des génomes », ainsi que les progrès de la recherche connexe dans les lois d'expansion et les modèles de génome.

HyperAI a compilé et résumé le merveilleux discours du Dr Li Mingchen sans en compromettre l'intention initiale. Voici une transcription des points saillants du discours.
Une nouvelle classification des modèles de langage des protéines : la relation entre la structure, la séquence et la fonction des protéines
Les protéines ont un large éventail d'applications, couvrant des domaines tels que le génie chimique, l'agriculture, l'alimentation, les cosmétiques, la médecine et les tests, avec une valeur marchande dépassant les milliards de dollars. En termes simples, la modélisation du langage des protéines est un problème de distribution de probabilités. Cela revient à déterminer la probabilité qu'une séquence d'acides aminés se produise dans la nature et à l'échantillonner en conséquence. Grâce à un pré-entraînement sur d'énormes quantités de données, le modèle peut représenter efficacement la distribution de probabilités observée dans la nature.
Le modèle de langage protéique a trois fonctions principales :
* Le processus d'apprentissage de la représentation de séquences protéiques sous forme de vecteurs de grande dimension
* Déterminer la rationalité de la séquence d'acides aminés
* Générer de nouvelles séquences protéiques
De nombreux articles de recherche classent les modèles de langage protéique selon l'architecture Transformer, les décrivant comme étant basés sur un codeur ou un décodeur Transformer. Cette classification est difficile à comprendre pour les chercheurs en biologie et est souvent source de confusion. C'est pourquoi je présenterai une nouvelle méthode de classification :Classification basée sur la relation entre la structure, la séquence et la fonction des protéines.
La séquence d'une protéine est sa séquence d'acides aminés. Une fois connue, elle peut être synthétisée en laboratoire ou en usine et appliquée en pratique. La structure d'une protéine est tout aussi cruciale. Sa fonction est due à sa structure spécifique dans l'espace tridimensionnel, ce qui lui permet de fonctionner à l'échelle microscopique.
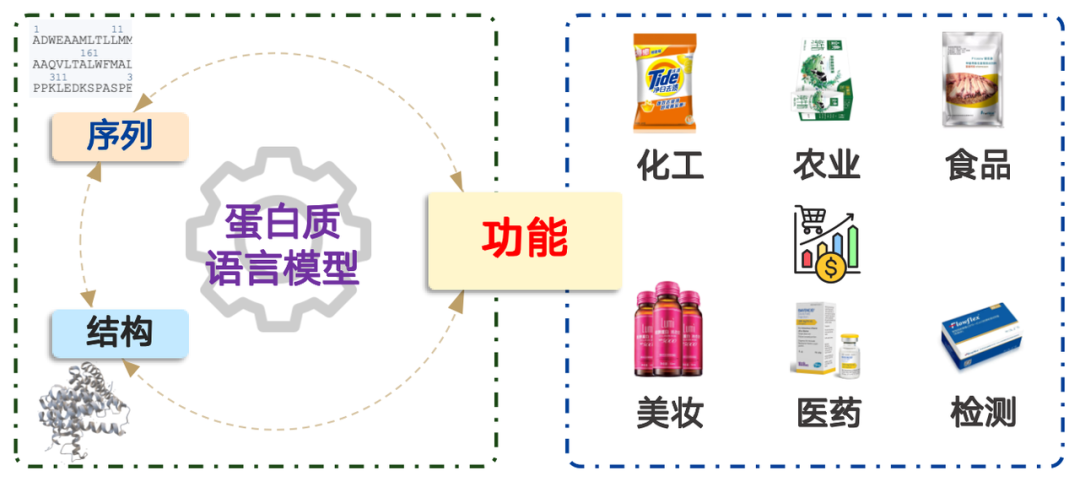
Sur la base de cette idée, les modèles de langage protéique peuvent être divisés en quatre catégories suivantes :
1. Séquence → Fonction :Prédire la fonction d'une séquence d'acides aminés donnée, c'est-à-dire Modèle de prédiction fonctionnelle.
2. Fonction → Séquence :Concevez la séquence d'acides aminés correspondante en fonction de la fonction donnée, y compris Modèles génératifset Modèle minier.
3. Séquence → Structure :La prédiction de sa structure en fonction de sa séquence d’acides aminés est généralement appelée « Modèle de prédiction structurelle »,Le modèle AlphaFold, lauréat du prix Nobel, appartient à ce type de modèle.
4. Structure → Séquence :La conception d’une séquence correspondante basée sur une structure protéique donnée est généralement appelée "Modèle de pliage inversé".
Scénarios d'application et voies techniques : Analyse de quatre modèles courants
« Séquence → Fonction »
La manière la plus simple de comprendre « séquence → fonction » est l’apprentissage supervisé.
Tout d'abord, le modèle de prédiction de fonction le plus basique consiste à exprimer des séquences protéiques sous forme de vecteurs, puis à les entraîner sur un jeu de données spécifique. Par exemple, pour prédire les points de fusion des protéines, il faut d'abord collecter un grand nombre d'étiquettes de points de fusion, convertir toutes les séquences protéiques de l'ensemble d'entraînement en vecteurs de grande dimension et les entraîner à l'aide de méthodes d'apprentissage supervisé. Enfin, il est possible d'inférer les séquences de l'ensemble de test ou de prédiction pour prédire la fonction. Cette approche, qui permet de gérer un large éventail de tâches, est un sujet de recherche en plein essor et permet d'obtenir des résultats relativement faciles.
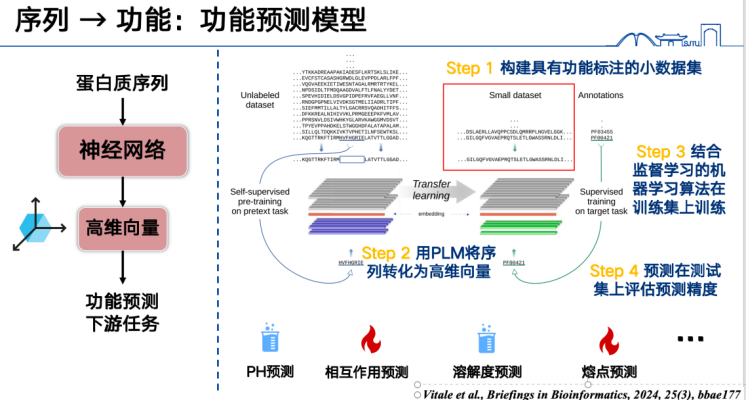
Deuxièmement, le modèle de langage protéique peut également prédire les fonctions de mutation.L’idée principale est d’apporter des modifications à certains acides aminés dans la séquence protéique, puis d’utiliser le modèle de langage protéique pour déterminer si le changement est « raisonnable ».
Le terme « raisonnable » ne fait pas ici référence à la conformité logique dans la vie réelle, mais plutôt à la conformité du changement d'acide aminé à la distribution de probabilité des séquences protéiques naturelles. Cette distribution de probabilité provient d'un grand nombre de statistiques réelles sur les séquences d'acides aminés, et ces distributions sont elles-mêmes le fruit de dizaines de millions d'années d'évolution.
Le modèle de langage protéique apprend ces lois évolutives lors de l'apprentissage et peut ainsi déterminer si une mutation est conforme ou non à ces lois. Mathématiquement, cette détermination peut être convertie en rapport des probabilités des deux séquences avant et après la mutation. Pour faciliter les calculs, ce rapport est souvent logarithmisé, ce qui le convertit en une forme de soustraction.
Le rapport de vraisemblance entre les protéines mutantes et les protéines sauvages utilisé par les modèles de langage permet d'estimer l'intensité de l'effet d'une mutation. Cette idée a été démontrée pour la première fois en 2018 dans un article de Nature Methods présentant le modèle DeepSequence, mais celui-ci était alors relativement petit. Par la suite, en 2021, le modèle ESM-1v a démontré que les modèles de langage protéique peuvent également prédire efficacement les effets des mutations grâce aux rapports de vraisemblance.
Pour évaluer la précision du modèle de prédiction de la fonction de mutation des protéines, une référence est nécessaire.
Les benchmarks sont de petits ensembles de données collectées pour mesurer la précision. Par exemple, ProteinGym, développé conjointement par la Harvard Medical School et l'Université d'Oxford, est le benchmark le plus couramment utilisé. Il contient des données sur 217 protéines mutantes et des millions de séquences de mutation. Les chercheurs attribuent des scores à chacune de ces séquences de mutation à l'aide d'un modèle de langage protéique, puis comparent les scores prédits du modèle aux scores réels. Une corrélation plus élevée indique une meilleure performance du modèle.
Cependant, ProteinGym est une référence à haut débit et à faible précision.Bien que limité par les conditions expérimentales, le modèle peut être testé à grande échelle, mais sa précision peut être limitée. La répétition d'une expérience peut entraîner des erreurs de corrélation entre les résultats et les données d'origine, ce qui fausserait les résultats de l'évaluation et les performances du modèle dans des applications réelles.
Pour résoudre ce problème,Nous avons développé un benchmark à faible débit et haute précision pour petits échantillons tel que VenusMutHub.Bien que la quantité de données ne soit pas importante, chaque élément de données est relativement précis et les résultats des expériences répétées sont presque cohérents, ce qui est plus proche des scénarios d'application réels.
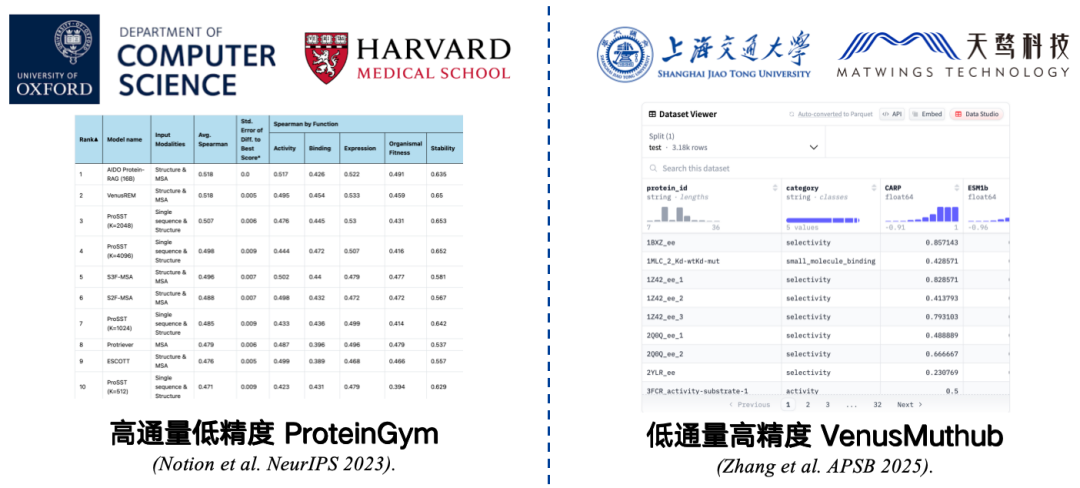
* Adresse du papier :Zhang L, Pang H, Zhang C, et al. VenusMutHub : Une évaluation systématique des prédicteurs de l'effet des mutations protéiques sur des données expérimentales à petite échelle[J]. Acta Pharmaceutica Sinica B, 2025, 15(5) : 2454-2467.
De plus, la structure peut être introduite pour améliorer la précision de la prédiction des mutations dans les modèles de langage protéique. L'année dernière, notre équipe a publié un article sur un modèle de langage protéique, le modèle ProSST, à NeurIPS. Ce modèle utilise à la fois des séquences d'acides aminés et des séquences structurées pour effectuer un pré-entraînement multimodal. ProSST s'est classé premier au ProteinGym Benchmark, le plus grand benchmark de prédiction des mutations à zéro coup.
* Adresse du papier :Li M, Tan Y, Ma X, et al. ProSST : Modélisation du langage des protéines avec structure quantifiée et attention démêlée[C]. Progrès dans les systèmes de traitement de l'information neuronale, 2024, 37 : 35700-35726.
Lorsque vous réalisez des expériences ou des conceptions, vous rencontrez souvent des questions telles que : « Quel modèle dois-je utiliser ? » et « Comment dois-je choisir en tant qu'utilisateur ? »
Dans une étude publiée cette année,Notre équipe a constaté que la perplexité du modèle de langage protéique pour la séquence cible peut refléter approximativement sa précision dans la tâche de prédiction de mutation.L'avantage est qu'il permet d'estimer les performances sans nécessiter de données de mutation de la protéine cible. Plus précisément, moins la perplexité est grande, meilleure est la compréhension de la séquence par le modèle, ce qui signifie souvent que ses prédictions de mutation pour cette séquence seront plus précises.
Sur la base de cette idée, nous avons développé un modèle d'ensemble, VenusEEM. Ce modèle pondère les modèles en fonction de leur perplexité ou sélectionne directement le modèle présentant la plus faible perplexité. Cela améliore considérablement la précision de la prédiction des mutations. Quelle que soit la stratégie choisie, le score de prédiction final reste relativement stable, ce qui évite une dégradation significative des performances due à une sélection incorrecte du modèle.
* Adresse du papier :Yu Y, Jiang F, Zhong B, et al. Sélection de modèles d'apprentissage profond à zéro coup pilotés par l'entropie pour les protéines virales[J]. Physical Review Research, 2025, 7(1) : 013229.
Enfin, dans le cadre de la recherche « de la séquence à la fonction », en plus des modèles mentionnés précédemment, notre équipe a également développé l'année dernière un nouveau modèle itératif de conception de mutations à site élevé, PRIME. Plus précisément, nous avons d'abord pré-entraîné un grand modèle de langage protéique sur 98 millions de séquences protéiques. Pour la tâche de prédiction des mutations à site élevé, nous avons d'abord obtenu des données de mutations à site faible et les avons introduites dans le modèle de langage protéique, les codant dans un vecteur de fonctions. À partir de ce vecteur de fonctions, nous avons ensuite entraîné un modèle de régression pour prédire les mutations à site élevé.Grâce à cette réaction itérative, un excellent produit protéique peut être développé en seulement 2 à 3 cycles d’expériences.
* Adresse papier:Jiang F, Li M, Dong J, et al. Un modèle de langage général guidé par la température pour concevoir des protéines de stabilité et d'activité améliorées[J]. Science Advances, 2024, 10(48) : eadr2641.

「Fonction→Séquence」
Nous avons déjà abordé la question de la déduction de la suite à la fonction. Voyons maintenant si nous pouvons inversement déduire la suite de la fonction.
Il existe un problème direct et inverse entre les suites et les fonctions. Alors que le problème direct consiste à trouver une réponse définitive, le problème inverse implique la recherche d'une solution soluble dans un vaste espace réalisable. Générer des suites à partir de fonctions constitue précisément ce problème inverse. En effet, les suites correspondent généralement à une ou plusieurs fonctions, mais une même fonction peut être implémentée par diverses suites complètement différentes. De plus, il n'existe pas de référence fiable pour le problème inverse. Lorsqu'un modèle génère des suites à partir d'une fonction donnée, sa précision ne peut généralement être testée qu'expérimentalement.

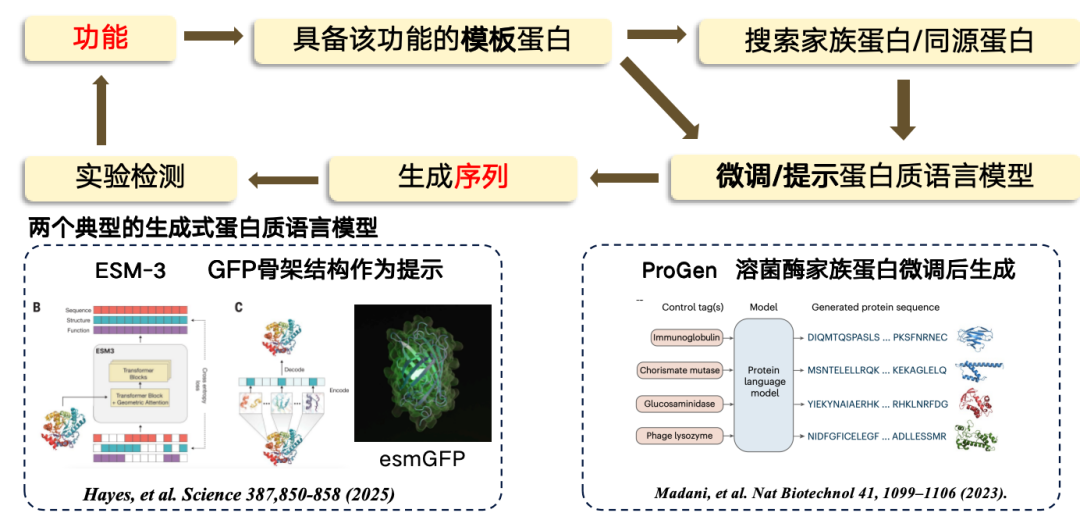
Dans l’étude actuelle,La conception, de la fonction à la séquence, adopte généralement une approche basée sur des modèles. Si une protéine modèle possède une fonction spécifique, elle peut servir de base pour trouver ou générer une nouvelle région.Le processus consiste d'abord à passer de la fonction à la séquence modèle, puis à rechercher certaines protéines familiales/protéines homologues à partir de la protéine modèle, puis à affiner le modèle de langage protéique, à utiliser le modèle de langage affiné pour générer de nouvelles régions de séquence, et enfin à effectuer des tests expérimentaux.
Actuellement, les deux modèles de langage protéique génératif les plus représentatifs comprennent :
*ESM-3, produit en utilisant la protéine fluorescente verte (GFP) comme modèle, mais la protéine résultante est moins fonctionnelle.
* ProGen est un modèle de langage purement autorégressif, similaire à ChatGPT, qui peut être généré à partir d'indices fonctionnels. Il est généré en affinant l'architecture protéique du lysozyme.
En plus de générer directement de nouvelles séquences protéiques,Vous pouvez également effectuer une recherche directement à partir de la quantité massive de séquences protéiques existantes.La protéine matrice est codée dans un espace de grande dimension, et la distance entre les vecteurs détermine si les deux protéines ont la même fonction. Enfin, les résultats sont extraits d'une base de données. Le principe de cette approche est que la distance entre les codages ou les vecteurs de deux protéines dans un espace de grande dimension peut refléter approximativement si les deux protéines ont des fonctions similaires.
La figure ci-dessous illustre deux exemples typiques d'exploration de modèles de langage protéique. Le premier est ESM-Ezy, développé par l'Université Westlake, qui utilise le modèle ESM-1b pour effectuer des recherches de vecteurs et extraire plusieurs expressions pour le remplissage. Le second est le modèle à grande échelle VenusMine, qui exploite des hydrolases TEP hautement performantes.
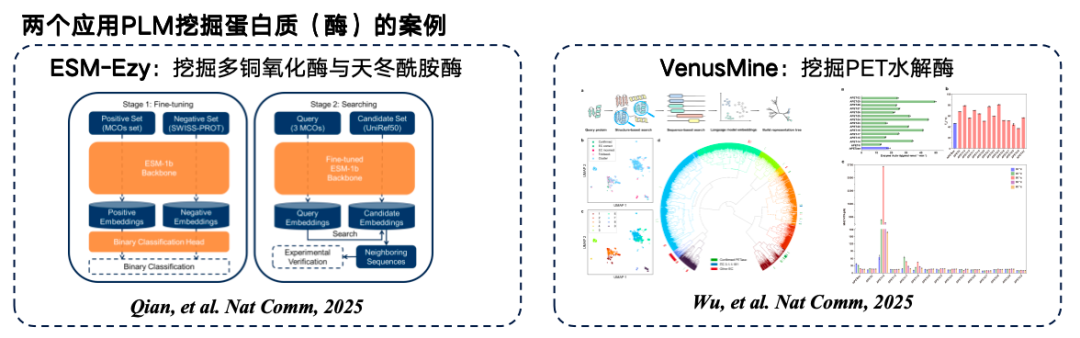
* Adresse papierWu B, Zhong B, Zheng L, et al. Exploiter le modèle de langage protéique pour la découverte basée sur la structure d'hydrolases TEP hautement efficaces et robustes[J]. Nature Communications, 2025, 16(1) : 6211.
En plus de « fonction → séquence », vous pouvez également ajouter « médiateur » entre la fonction et la séquence :
* Lorsque la structure est utilisée comme intermédiaire : la structure de la protéine est déduite en fonction de la fonction (outils courants tels que la diffusion RF), et la structure générée est ensuite entrée dans un modèle de langage de repliement inverse des protéines (tel que ProteinMPNN) pour finalement générer une séquence.
* Lorsque le langage naturel est utilisé comme support : par exemple, la méthode décrite dans l'article de recherche « Un cadre de conception de protéines guidée par le texte » aligne le langage naturel et les séquences protéiques dans un espace de haut niveau grâce à l'apprentissage comparatif. Une séquence protéique peut ensuite être générée directement dans cet espace de haut niveau grâce au guidage en langage naturel.
Séquence → Structure
Dans le sens séquence-structure, le modèle le plus classique est sans conteste AlphaFold. Alors, pourquoi avons-nous encore besoin de modèles de langage protéique pour prédire la structure ?La raison principale est la rapidité.
La principale raison de la lenteur d'AlphaFold est que sa recherche MSA (alignement de séquences multiples) repose sur le processeur pour interroger de grandes bases de données. Si l'accélération GPU est possible, l'accélération réelle est encore plus lente. De plus, AlphaFold nécessite une correspondance de modèles pendant le processus de repliement, ce qui est également très chronophage. Remplacer ces deux modules par un modèle de langage protéique peut considérablement accélérer le processus de prédiction de structure. Cependant, selon les recherches actuellement publiées, la précision de la prédiction de structure basée sur les modèles de langage protéique reste généralement inférieure à celle du modèle AlphaFold sur la plupart des indicateurs d'évaluation.
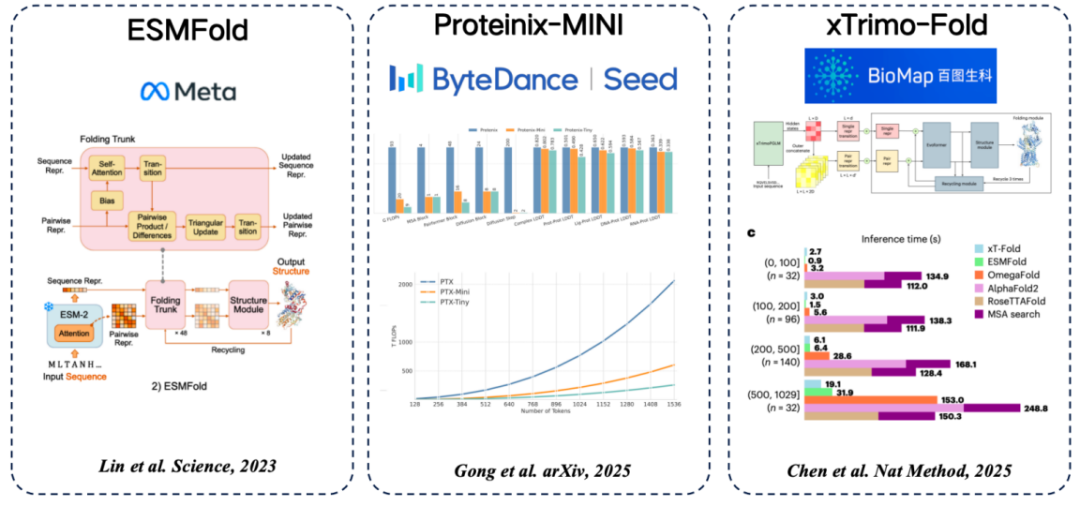
Il existe plusieurs modèles courants de langage protéique, de la séquence à la structure.L'idée commune d'utiliser des caractéristiques extraites de modèles de langage protéique pour remplacer MSA est adoptée :
* ESMFold (Meta) : la première méthode permettant de prédire directement la structure des protéines à l'aide d'un modèle de langage protéique, permettant d'obtenir une grande précision sans recourir à la recherche MSA.
* Proteinix-MINI (ByteDance) : utilise le modèle de langage protéique au lieu de MSA, qui permet également d'obtenir des résultats très rapides et a une précision de prédiction proche du modèle AlphaFold 3.
* xTrimo-Fold (Baidu Biosciences) : il utilise les fonctionnalités d'un modèle à 100 milliards de paramètres au lieu de MSA, ce qui accélère la recherche.
Structure → Séquence
La structure est conçue sur la base de fonctions connues, mais comment la synthétiser en laboratoire ?Nous devons également le convertir en une séquence d’acides aminés, ce qui constitue le « modèle de langage de repliement inverse » mentionné précédemment.
Le modèle de langage de repliement inverse peut être considéré comme le « problème inverse » d'AlphaFold. Contrairement à AlphaFold, qui prédit la structure 3D à partir des séquences d'acides aminés, le modèle de repliement inverse vise à apprendre une fonction de mappage de la structure 3D d'une protéine à sa séquence d'acides aminés.
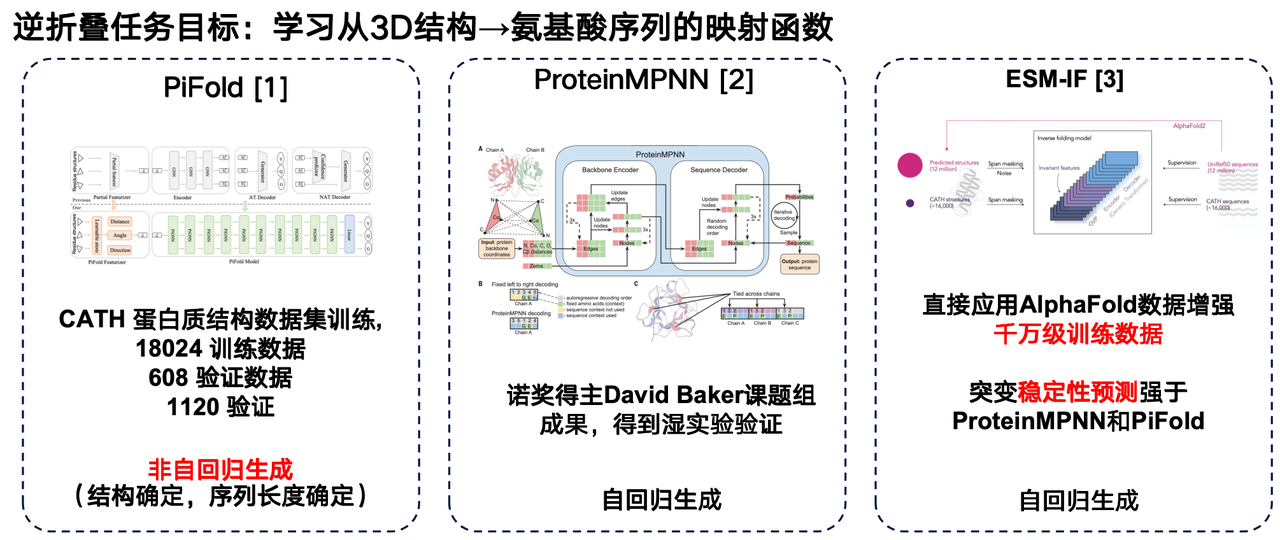
Je souhaite partager plusieurs travaux dans ce domaine : le premier concerne le modèle PiFold de l'équipe de recherche de l'Université Westlake. Une innovation majeure de son architecture réside dans l'utilisation d'une méthode de génération non autorégressive.
Le second est ProteinMPNN, développé par le groupe de recherche de David Baker. Ce modèle de repliement inverse, l'un des plus utilisés, utilise la génération autorégressive pour coder les structures protéiques individuelles via des réseaux neuronaux graphiques, puis génère les séquences d'acides aminés une par une.
L'ESM-IF de Meta constitue également une avancée significative. Son point fort réside dans l'exploitation des données structurelles massives prédites par AlphaFold pour prédire uniformément les structures tridimensionnelles correspondantes de dizaines de millions de séquences protéiques, constituant ainsi un ensemble d'apprentissage extrêmement vaste. Les données d'apprentissage d'ESM-IF atteignent des dizaines de millions d'unités et le nombre de paramètres du modèle dépasse les 100 millions. Grâce à cela, le modèle effectue non seulement des tâches de repliement inverse, mais démontre également d'excellentes performances dans la prédiction de la stabilité mutationnelle.
Plusieurs approches pour améliorer les modèles de langage des protéines
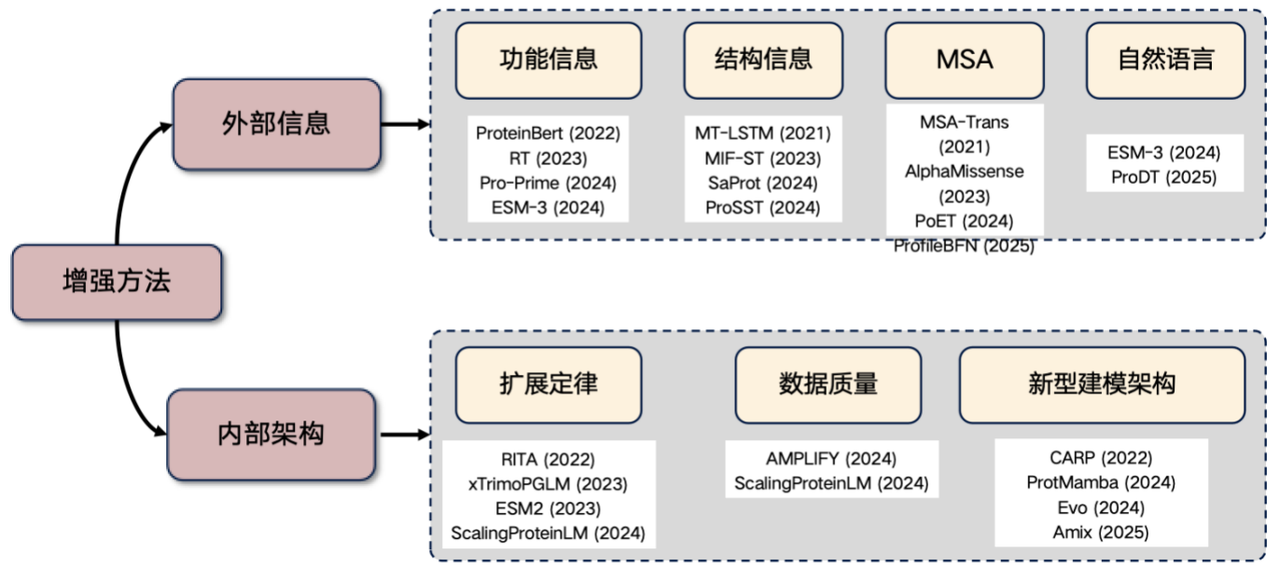
Enfin, permettez-moi d'ajouter un axe de recherche très populaire actuellement : l'amélioration du modèle de langage des protéines. Si vous envisagez de mener des recherches dans ce domaine, vous pouvez partir des idées suivantes :Introduire des informations externes et améliorer la structure interne.
1. Présentation des informations externes
* Informations fonctionnelles : Par exemple, saisie de caractéristiques telles que la température et le pH dans le transformateur. Ces informations peuvent être intégrées explicitement aux données d'entrée du modèle ou par des actions apprises afin d'améliorer les performances du modèle de langage protéique.
* Informations structurelles : Présentation d'informations sur la structure tridimensionnelle ou la séquence structurée.
* Informations MSA : l'alignement de séquences multiples (MSA) est un type d'information très utile. Son intégration au modèle de langage peut souvent améliorer considérablement les performances.
* Informations en langage naturel : Ces dernières années, certaines études ont tenté d’intégrer des informations en langage naturel, mais cette direction est encore à l’étude.
2. Améliorer l'architecture interne
* Loi d'échelle : l'amélioration des performances est obtenue en augmentant considérablement le nombre de paramètres du modèle et la taille des données d'entraînement.
* Améliorer la qualité des données : réduire le bruit dans les données et améliorer la précision.
* Exploration de nouvelles architectures : telles que les architectures CARP, ProtMamba et Evo.
Ces dernières années, l’utilisation des informations sur la structure des protéines pour améliorer les performances des modèles est devenue une direction de recherche en vogue.
L'une des premières études représentatives est l'article de 2021 « Learning the protein language: Evolution, structure, and function », qui a démontré comment l'information structurelle peut être utilisée pour améliorer les capacités des modèles de langage protéique. Par la suite, le modèle SaProt a proposé une approche astucieuse : il concatène le vocabulaire des acides aminés de la protéine avec 20 vocabulaires de structures virtuelles générés par Foldseek pour les structures protéiques, générant ainsi un vocabulaire combiné de 400 (20 × 20) mots. Ce vocabulaire a été utilisé pour entraîner un modèle de langage masqué, avec une excellente précision.
Notre équipe a également entraîné de manière indépendante un modèle de pré-entraînement multimodal, ProSST, pour la séquence et la structure des protéines. Ce modèle permet une représentation discrète des informations structurelles en convertissant la structure continue de la protéine en 2 048 jetons distincts.
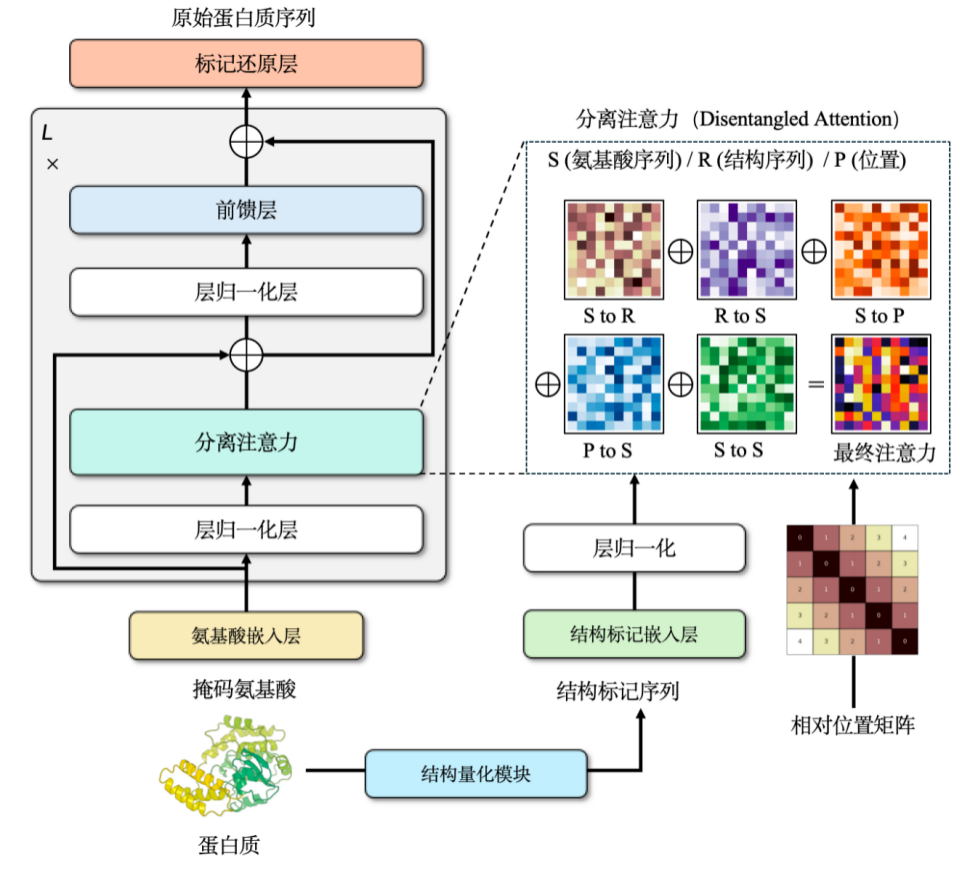
L'intégration d'informations structurelles dans les modèles de langage protéique peut améliorer considérablement leurs performances. Cependant, un problème peut survenir lors de ce processus : si les données structurelles prédites par AlphaFold sont directement utilisées pour l'apprentissage, alors que la perte sur l'ensemble d'apprentissage diminue progressivement, celle sur l'ensemble de validation ou l'ensemble de test augmente progressivement.La clé pour résoudre ce problème est de régulariser les informations structurelles.En termes simples, cela signifie simplifier des données complexes pour les rendre plus adaptées au traitement des modèles.
Les structures protéiques sont généralement représentées par des coordonnées continues dans un espace tridimensionnel. Il est nécessaire de simplifier cette représentation en les convertissant en séquences discrètes d'entiers. Pour ce faire, nous avons utilisé une architecture de réseau neuronal graphique et l'avons entraînée avec un encodeur de débruitage, ce qui a permis de construire un vocabulaire de structures discrètes d'environ 2 048 jetons.
Avec des informations structurelles et séquentielles,Nous avons choisi le mécanisme d’attention croisée pour combiner les deux.Cela permet au modèle Transformer modifié d'intégrer à la fois des séquences d'acides aminés et des séquences structurales. Lors de la phase de pré-apprentissage, nous avons conçu ce modèle comme une tâche de développement de modèle de langage.Les données de formation contiennent plus de 18,8 millions de structures protéiques de haute qualité avec une taille de paramètre d'environ 110 millions.Le modèle a obtenu des résultats de pointe à l'époque, et bien qu'il ait depuis été surpassé par des modèles plus récents, il détenait toujours les meilleurs résultats de sa catégorie au moment de sa publication.
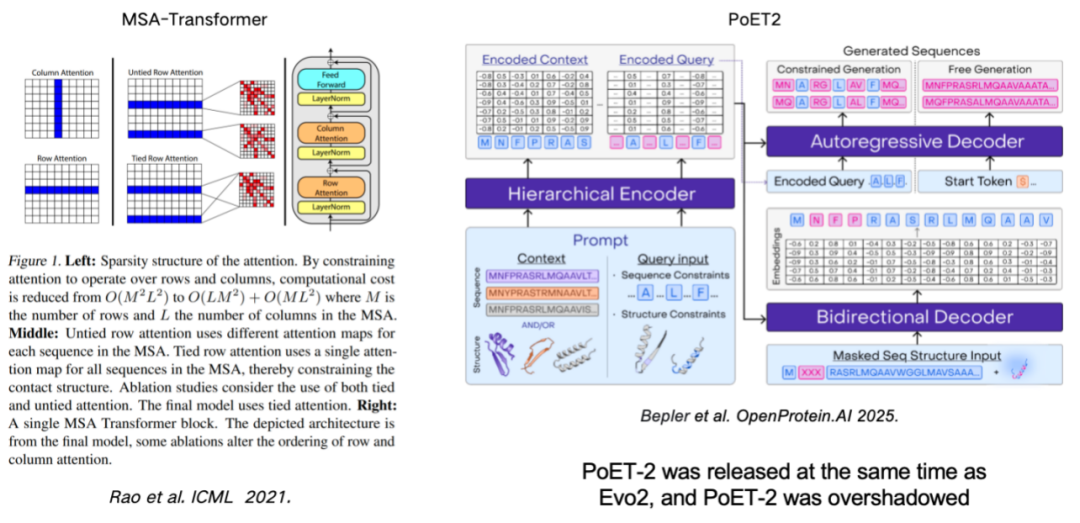
L’utilisation de l’AMS (alignement de séquences multiples) pour améliorer le modèle de langage protéique est également un moyen important d’améliorer les performances du modèle.Ces travaux remontent au MSA-Transformer, qui a efficacement intégré les informations MSA au modèle en introduisant des règles de lignes et de colonnes. Le modèle PoET2, récemment publié, utilise un encodeur hiérarchique pour traiter les informations MSA et les intégrer dans une architecture de modèle passe-partout. Après un entraînement à grande échelle, il a démontré des performances exceptionnelles.
La loi de l’échelle : un modèle plus grand est-il toujours plus puissant ?
La loi dite d'échelle est issue du traitement automatique du langage naturel. Elle révèle une loi universelle :Les performances du modèle continueront de s’améliorer avec l’augmentation de l’échelle des paramètres, du volume des données de formation et des ressources de calcul.
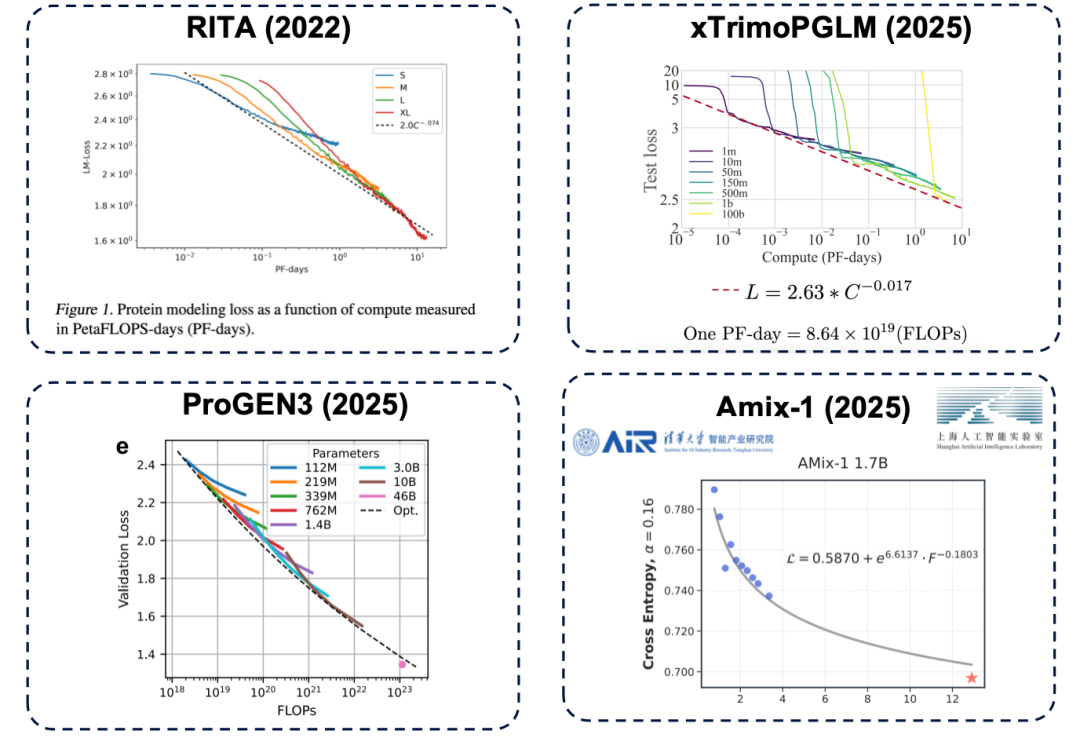
La taille des paramètres est un facteur clé pour déterminer la limite supérieure des performances d'un modèle. Si le nombre de paramètres est insuffisant, même en investissant davantage de ressources de calcul (en termes simples, en dépensant plus d'argent), les performances du modèle atteindront un goulot d'étranglement. Ce même principe existe également dans le domaine des modèles de langage protéique et a été confirmé par de nombreuses études, notamment des travaux représentatifs tels que RITA, xTrimoPGLM, ProGEN3 et Amix-1.
* Modèle RITA : développé par l'Université d'Oxford, la Harvard Medical School et LightOn AI.
* Modèle xTrimoPGLM : Développé par l'équipe Baitu Bioscience, il met à l'échelle les paramètres du modèle à environ 100 milliards.
* Modèle ProGEN3 : développé par l'équipe Profluent Biotech.
* Modèle Amix-1 : Proposé par l'Institut des industries intelligentes de l'Université Tsinghua et le Laboratoire d'intelligence artificielle de Shanghai, il utilise une architecture de réseau de correspondance de flux bayésienne et dispose également d'une loi d'expansion.
La « loi d'échelle » mentionnée précédemment fait référence au processus de pré-apprentissage. Cependant, en recherche sur les protéines, c'est la performance des tâches en aval qui nous intéresse en fin de compte. Ceci soulève la question suivante :L’amélioration des performances avant la formation aide-t-elle nécessairement les tâches en aval ?
Dans l'évaluation xTrimoPGLM, l'équipe de recherche a constaté que dans environ 441 tâches en aval TP3T, il existe effectivement une corrélation positive entre « de meilleures performances avant l'entraînement et des performances en aval plus fortes ».
Parallèlement, le modèle Amix-1 a démontré une capacité émergente dans les tâches de prédiction de structure. Il s'agit de tâches où un petit modèle est totalement incapable de résoudre le problème, mais où les performances s'améliorent soudainement et significativement lorsque la taille des paramètres du modèle dépasse un certain seuil critique. Dans cette expérience, ce phénomène était particulièrement prononcé dans les tâches de prédiction de structure, où l'amélioration des performances a montré une « ligne rouge en forme de falaise » lorsque la taille des paramètres dépassait le seuil critique.
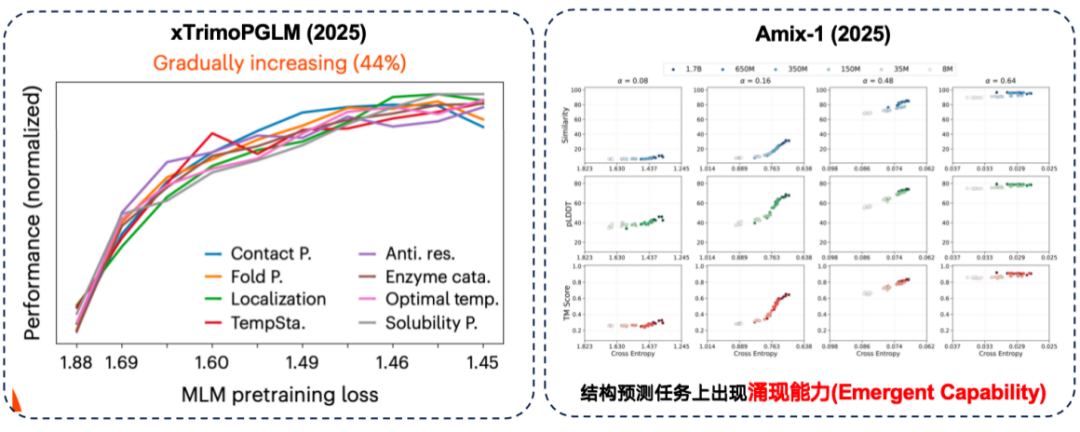
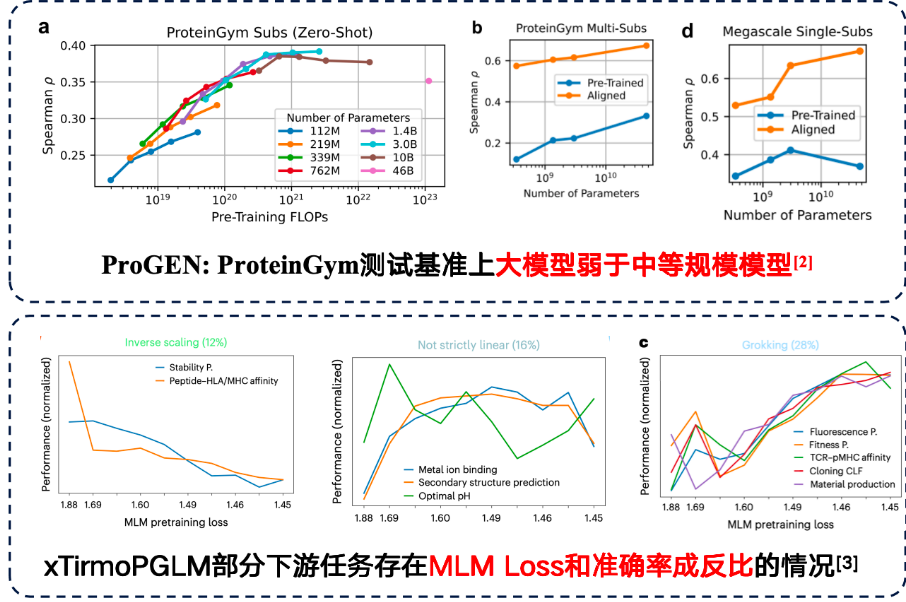
Bien que dans certaines tâches, les grands modèles puissent effectivement apporter de meilleures performances en aval.Cependant, les tâches en aval ont également découvert une loi d’échelle inverse.Autrement dit, plus le modèle est petit, meilleures sont les performances.
Des études ont montré que la simple augmentation du nombre de paramètres du modèle n'améliore pas les résultats lorsque les données d'entraînement sont elles-mêmes très bruitées. La qualité des données doit donc faire l'objet d'une attention accrue. Dans la tâche de prédiction des mutations protéiques du benchmark ProteinGym, les modèles de taille moyenne ont même obtenu de meilleurs résultats en termes de précision. De plus, l'équipe développant xTirmoPGLM a également découvert des cas de corrélation non positive, où les performances avant l'entraînement ne correspondaient pas aux performances de la tâche en aval.
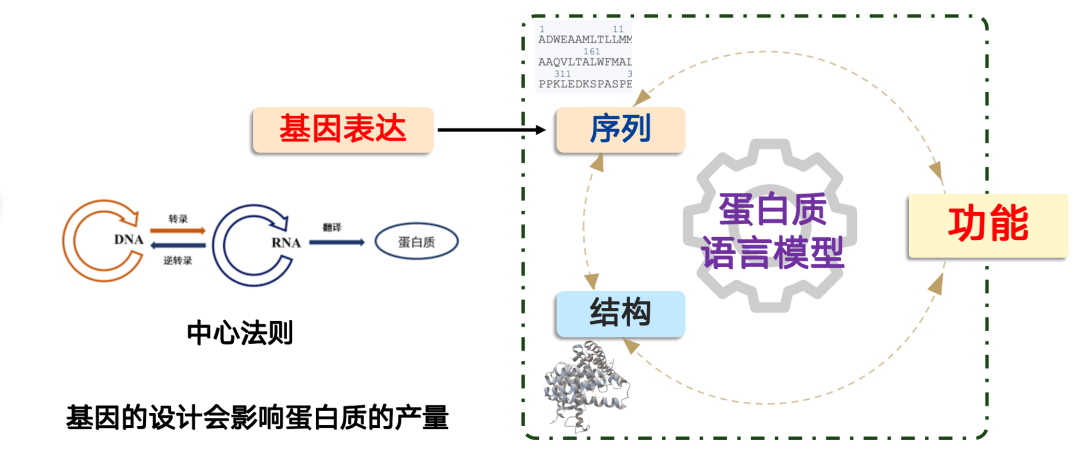
Modélisation génomique : de la conception de l'ADN à l'optimisation du rendement protéique
Le problème que résout le modèle du génome est : comment produisons-nous une protéine ?
En biologie synthétique,La production d'une protéine suit le dogme central de la biologie moléculaire : « ADN → ARN → protéine ».Dans les cellules, ce processus est contrôlé par le corps cellulaire, et nous pouvons le compléter en concevant des gènes. Mais l'essentiel est que la conception des gènes affecte directement la production de protéines.
Dans les applications pratiques, nous rencontrons souvent des situations où une protéine présente d'excellentes performances fonctionnelles, mais où, en raison d'une conception génétique défaillante, son niveau d'expression est extrêmement faible, ce qui ne permet pas de répondre aux besoins d'industrialisation ou d'application à grande échelle. Dans ce cas, les modèles d'IA peuvent jouer un rôle.
La mission du modèle d'IA est de déduire comment concevoir des séquences d'ADN directement à partir de séquences protéiques et d'en accroître la production. Le modèle proposé par notre équipe, ProDMM, repose sur une stratégie de pré-apprentissage et se compose de deux phases :
Dans la première phase, un pré-entraînement conjoint est utilisé pour apprendre les représentations des protéines et de l'ADN. Les entrées comprennent des séquences de protéines et d'ADN, et un modèle de langage est entraîné à l'aide d'une architecture Transformer. L'objectif est d'apprendre simultanément les représentations des protéines, des codons et des séquences d'ADN. Dans la deuxième phase, des tâches génératives sont entraînées sur des tâches en aval, comme la conversion de la protéine à la séquence codante (CDS). Étant donnée une protéine, une séquence d'ADN peut être générée.
* Adresse du papier :Li M, Ren Y, Ye P, et al. Exploiter une modélisation de séquence multimodale unifiée pour dévoiler l'interdépendance protéine-ADN[J]. bioRxiv, 2025 : 2025.02. 26.640480.
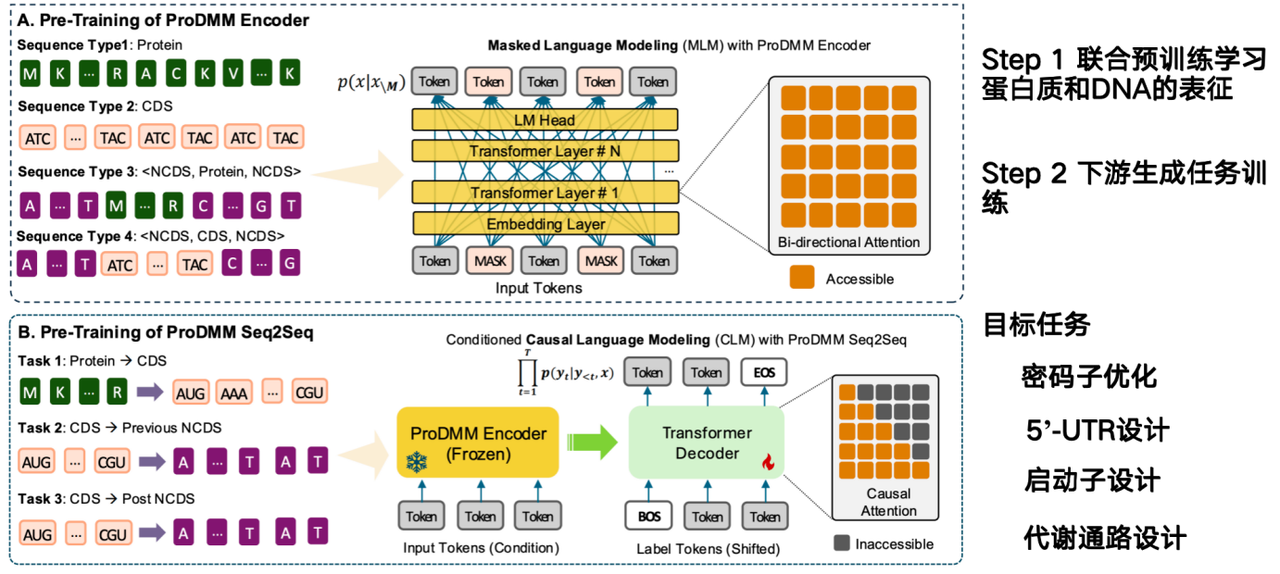
L'objectif du projet des codons à l'ADN non codant (NCDS) est de compléter l'optimisation des codons, la conception 5'-UTR, la conception du promoteur et la conception de la voie métabolique.
La conception des voies métaboliques implique le travail coordonné de plusieurs protéines au sein d'un gène pour synthétiser un produit spécifique. Nous devons optimiser les produits de l'ensemble de la voie métabolique, une tâche particulièrement adaptée aux modèles génomiques, car les modèles protéiques n'optimisent que pour une seule protéine et sont indépendants du contexte. Cependant, un défi majeur pour les modèles génomiques réside dans la nécessité de prendre en compte les interrelations au sein de l'environnement cellulaire, ce qui constitue actuellement leur plus grand défi.
À propos du Dr Li Mingchen
L'intervenant invité de cette séance de partage est Li Mingchen, chercheur postdoctoral au sein du groupe de recherche de Hong Liang à l'Institut des sciences naturelles de l'Université Jiao Tong de Shanghai. Il est titulaire d'un doctorat en ingénierie informatique et technologique et d'une licence en mathématiques de l'Université des sciences et technologies de Chine orientale. Ses recherches portent principalement sur le pré-entraînement et le perfectionnement des modèles de langage des protéines.
Il a remporté le titre de Shanghai Outstanding Graduate, la bourse nationale et la médaille d'or de la division de Shanghai du concours « Internet+ » pour l'innovation et l'entrepreneuriat des étudiants universitaires. Il a publié dix articles scientifiques en tant que premier auteur, co-premier auteur ou auteur correspondant dans des revues et conférences telles que NeurIPS, Science Advances, Journal of Chimioformatics et Physical Review Research, et a participé à la publication de dix articles scientifiques.
Obtenez des articles de haute qualité et des articles d'interprétation approfondis dans le domaine de l'IA4S de 2023 à 2024 en un seul clic⬇️









