Command Palette
Search for a command to run...
L'activité Des Variantes Protéiques Conçues a Été Multipliée Par 50 ! L'équipe Zhou Hao De l'AIR De l'Université Tsinghua a Proposé AMix-1 Basé Sur Des Réseaux De Flux Bayésiens Pour Obtenir Une Conception De Protéines Évolutive Et universelle.

Actuellement, la recherche dans le domaine des modèles de piédestal protéique reste bloquée à l'ère du « BERT », incapable de s'adapter pleinement aux propriétés biologiques des séquences protéiques. Auparavant, des modèles d'IA tels qu'AlphaFold et ESM ont considérablement progressé dans de nombreux domaines, notamment la prédiction de structure, le repliement inverse, la prédiction des propriétés fonctionnelles, l'évaluation des effets des mutations et la conception des protéines.Cependant, ces modèles manquent encore de méthodologies évolutives et systématiques similaires aux modèles de langage à grande échelle (LLM) de pointe, et leurs capacités ne peuvent pas être continuellement améliorées avec l'augmentation du volume de données, de l'échelle du modèle et des ressources de calcul.
Le manque d'universalité de ces modèles a entraîné des défis difficiles à résoudre dans le domaine de la conception des protéines : les modèles ne peuvent pas capturer l'hétérogénéité conformationnelle des protéines, et les prédictions sur la conception des protéines ne peuvent pas aller au-delà de la portée des données de formation ; et la dépendance excessive au transfert des méthodologies NLP a conduit à l'absence de conceptions architecturales originales ciblant les caractéristiques des protéines.
Dans ce contexte, le groupe de recherche de Zhou Hao à l'Institut des industries intelligentes (AIR) de l'Université Tsinghua, en collaboration avec le Laboratoire d'intelligence artificielle de Shanghai, a proposé un modèle de base de protéines AMix-1 systématiquement formé basé sur un réseau de flux bayésien, offrant un chemin évolutif et général pour la conception de protéines.Ce modèle a adopté pour la première fois la méthodologie systématique de la « loi de mise à l'échelle de pré-entraînement », de la « capacité émergente », de l'« apprentissage en contexte » et de la « mise à l'échelle au moment du test », et a conçu une stratégie d'apprentissage contextuel basée sur l'alignement de séquences multiples (MSA) sur cette base, obtenant une cohérence dans le cadre général de la conception des protéines tout en garantissant l'évolutivité du modèle.
Les résultats de recherche pertinents ont été publiés sur la plateforme arXiv sous le titre « AMix-1 : A Pathway to Test-Time Scalable Protein Foundation Model ».
Points saillants de la recherche :
* Une loi d’échelle prévisible a été établie pour le modèle de génération de protéines basé sur le réseau de flux bayésien ;
* Le modèle AMix-1 développe spontanément une « compréhension perceptive » de la structure des protéines grâce à des objectifs de formation au niveau de la séquence uniquement, sans nécessiter de supervision structurelle explicite.
* Le cadre d'apprentissage contextuel basé sur l'alignement de séquences multiples (MSA) résout le problème d'alignement dans l'optimisation fonctionnelle, améliore les capacités de raisonnement et de conception du modèle dans un contexte évolutif et permet à AMix-1 de générer de nouvelles protéines avec une structure et une fonction conservées ;
* Proposer un algorithme d'extension du temps de test guidé par les coûts de vérification pour permettre une nouvelle approche de conception basée sur l'évolution lorsque les budgets de vérification augmentent.
Adresse du document :
Suivez le compte officiel et répondez « AMix » pour obtenir le PDF complet
Autres articles sur les frontières de l'IA :
Ensemble de données UniRef50 : prétraitement et clustering itératif
Les chercheurs ont utilisé le jeu de données UniRef50 prétraité lors du pré-apprentissage du modèle. Ce jeu de données, fourni par EvoDiff, est dérivé d'UniProtKB et filtré à partir des séquences UniParc par clustering itératif (UniProtKB+UniParc → UniRef100 → UniRef90 → UniRef50)., contenant 41 546 293 séquences d'apprentissage et 82 929 séquences de validation. Les séquences de plus de 1 024 résidus ont été réduites à 1 024 résidus grâce à une stratégie d'élagage aléatoire afin de réduire les coûts de calcul et de générer des sous-séquences diversifiées. Ce processus itératif garantit une représentation de haute qualité, non redondante et diversifiée des séquences UniRef50, offrant une couverture étendue de l'espace des séquences protéiques pour les modèles de langage protéique.
Téléchargez le jeu de données UniRef50 :
Solutions techniques systématiques
AMix-1 fournit un ensemble complet de solutions techniques systématiques pour la mise en œuvre de la mise à l'échelle du temps de test pour les modèles de piédestal protéique :
* Loi d'échelle de pré-formation :Il est clair comment équilibrer les paramètres, le nombre d’échantillons et l’effort de calcul pour maximiser les capacités du modèle ;
* Capacité émergente :Cela montre qu’au fur et à mesure que la formation progresse, le modèle émergera avec une « compréhension perceptive » de la structure des protéines ;
* Apprentissage en contexte :Il résout le problème d'alignement dans l'optimisation fonctionnelle, permettant au modèle d'apprendre le raisonnement et la conception dans un contexte évolutif ;
* Mise à l'échelle du temps de test :AMix-1 ouvre une nouvelle approche de la conception basée sur l'évolution à mesure que les budgets de vérification augmentent.
De la formation et de l'inférence à la conception, AMix-1 a démontré sa polyvalence et son évolutivité en tant que modèle de base de protéines, ouvrant la voie à une mise en œuvre pratique.
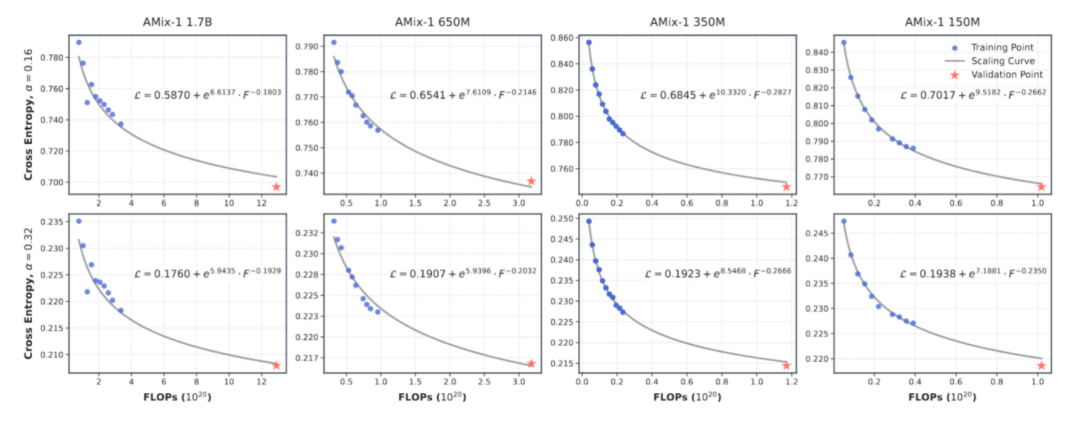
Loi d'échelle de pré-entraînement : capacités prévisibles des modèles protéiques
Pour obtenir une loi d'échelle prévisible pour AMix-1, cette étude a conçu une combinaison de modèles multi-échelles avec des paramètres allant de 8 millions à 1,7 milliard dans l'expérience, et a utilisé des opérations à virgule flottante d'entraînement (FLOP) comme indicateur de mesure unifié pour ajuster et prédire avec précision la relation de loi de puissance entre la perte d'entropie croisée du modèle et la quantité de calcul.
À en juger par les résultats, la courbe de puissance entre la perte du modèle et l’effort de calcul est très cohérente, confirmant que le processus de formation du modèle basé sur le réseau de flux bayésien est hautement prévisible.
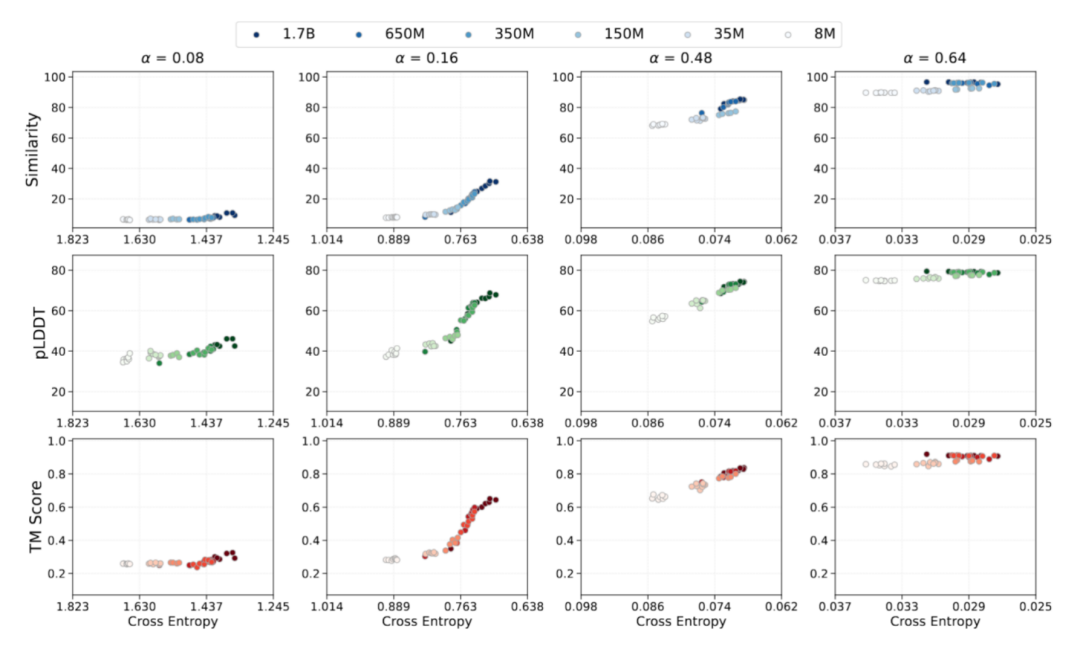
Capacité émergente : atteindre des capacités de modèle avancées
Dans l'apprentissage des séquences protéiques, l'étude de l'émergence structurale repose généralement sur le paradigme « séquence-structure-fonction ». Afin de valider le lien entre la dynamique d'optimisation et les résultats fonctionnels en modélisation protéique, l'équipe de recherche a analysé le comportement émergent selon une approche centrée sur les pertes, basée sur une loi d'échelle prévisible. En utilisant la perte d'entropie croisée prédictive comme point d'ancrage, ils ont empiriquement associé la perte d'apprentissage à la performance de génération de protéines. L'évaluation de la capacité d'émergence du modèle, réalisée dans le cadre de cette étude, s'est concentrée sur trois aspects :
* La capacité du modèle à récupérer les niveaux de séquence à partir de distributions de séquences corrompues sur la base d’observations de cohérence de séquence ;
* La transition des modèles de la compréhension séquentielle à la faisabilité structurelle du point de vue de la pliabilité ;
* Juger la capacité du modèle à maintenir les caractéristiques structurelles à partir de la cohérence structurelle.
Les données pertinentes lors de la formation AMix-1 démontrent pleinement le processus d'émergence des capacités de « cohérence de séquence, de repliabilité et de cohérence structurelle » du modèle de base protéique.Les données montrent que tous les indicateurs de capacité du modèle pendant la formation sont fortement corrélés avec la perte d'entropie croisée, ce qui vérifie la possibilité de prédire la capacité du modèle via la loi d'échelle et la perte d'entropie croisée.Dans le même temps, lorsqu'il est formé uniquement avec des objectifs auto-supervisés au niveau de la séquence et sans introduire aucune information structurelle, le modèle présente toujours une capacité d'urgence après que la perte d'entropie croisée tombe à un seuil, montrant une transition non linéaire entre pLDDT et TM-score.
Apprentissage en contexte : un paradigme général pour la conception des protéines
Grâce à des simulations informatiques, les chercheurs ont vérifié le mécanisme d'apprentissage contextuel d'AMix-1. Les expériences de simulation ont montré queAMix-1 est capable d'extraire et de généraliser avec précision les contraintes structurelles ou fonctionnelles à partir d'échantillons d'entrée sans s'appuyer sur des étiquettes explicites ou une supervision structurelle.
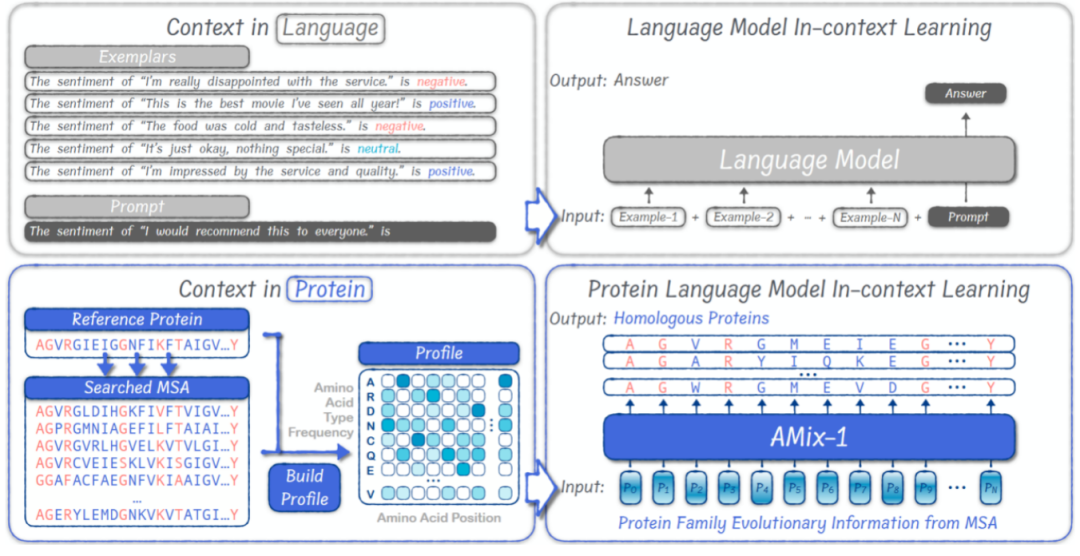
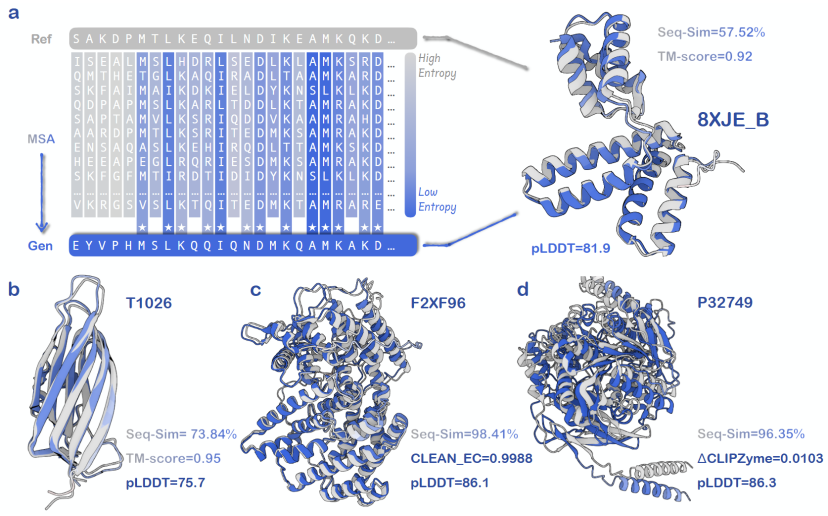
Comparé à la conception protéique traditionnelle, qui nécessite des processus personnalisés en fonction des types de tâches et manque d'un cadre unifié, AMix-1 introduit un mécanisme d'apprentissage en contexte (ICL) au sein d'un vaste modèle de langage pour réaliser une conception protéique guidée par la structure et la fonction. Des expériences ont montré que, dans les tâches structurelles, AMix-1 peut générer de nouvelles protéines aux structures prédites très cohérentes en utilisant comme repères des protéines homologues conventionnelles, voire des protéines pratiquement sans homologie. Dans les tâches fonctionnelles, AMix-1 peut générer des protéases très cohérentes en fonction de la fonction enzymatique et de la conception guidée par la réaction chimique de la protéine d'entrée.
Dans le cadre de ce mécanisme général,Le modèle peut déduire automatiquement les informations et les règles communes à un groupe donné de protéines et utiliser ces règles pour guider la génération de nouvelles protéines conformes aux règles communes.Ce mécanisme compresse un groupe de MSA protéiques dans une distribution de probabilité au niveau de la position (profil) entrée dans le modèle. Après une analyse rapide de la structure et des règles fonctionnelles des protéines d'entrée, le modèle peut générer de nouvelles protéines répondant à l'objectif.
Mise à l'échelle du temps de test : intelligence générale évolutive
En s'appuyant sur l'approche de mise à l'échelle du temps de test, les chercheurs ont utilisé le framework Proposer-Verifier pour développer EvoAMix-1. En augmentant continuellement le budget de vérification, ils ont amélioré les performances du modèle d'AMix-1. Tout en améliorant l'efficacité de conception du modèle, l'équipe a également atteint l'évolutivité. De plus, pour garantir la compatibilité, l'équipe a supprimé les exigences prédéfinies pour les propriétés du vérificateur.
EvoAMix-1 favorise l'exploration basée sur le caractère aléatoire inhérent aux modèles probabilistes. En intégrant des fonctions de récompense issues de simulations informatiques spécifiques à chaque tâche ou des retours de détection expérimentale, il génère et sélectionne de manière itérative des séquences protéiques candidates sous contraintes évolutives. Il permet une évolution protéique dirigée efficace sans ajustement fin du modèle, offrant ainsi des performances robustes et évolutives lors des tests de conception protéique.Dans les six tâches de conception, EvoAMix-1 surpasse systématiquement AMix-1 dans l'apprentissage en contexte et diverses méthodes de base solides.
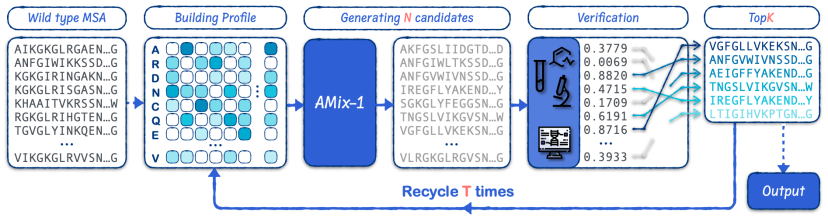
Par rapport à la méthode traditionnelle de génération de nouvelles variantes de protéines par échantillonnage d'importance,EvoAMix-1 ne met pas à jour les paramètres du modèle, mais construit plutôt une distribution de propositions via des exemples contextuels.À chaque tour, AMix-1 prend comme indices un ensemble d'alignements de séquences multiples (MSA) ou leurs spectres, qui sont considérés comme des conditions d'entrée pour un modèle de base protéique, qui échantillonne ensuite les séquences voisines, définissant ainsi efficacement une nouvelle distribution de propositions conditionnelles.
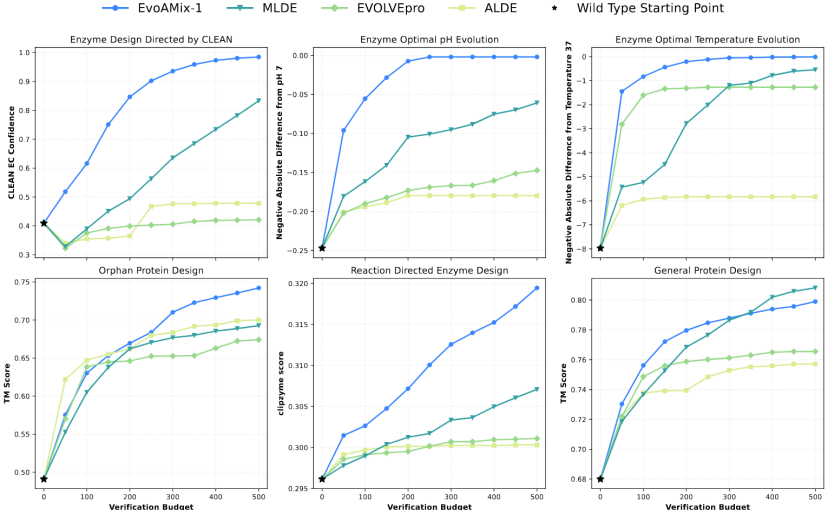
L'équipe de recherche a validé systématiquement la polyvalence et l'évolutivité d'EvoAMix-1 dans plusieurs tâches représentatives d'évolution dirigée vers les protéines, notamment l'évolution optimale du pH et de la température des enzymes, la préservation et l'amélioration des fonctions, la conception de protéines orphelines et l'optimisation générale guidée par la structure. Les résultats expérimentaux démontrent la robustesse de l'évolutivité de la mise à l'échelle d'EvoAMix-1 en fonction du temps de test, démontrant ainsi sa grande polyvalence entre les tâches et les objectifs.
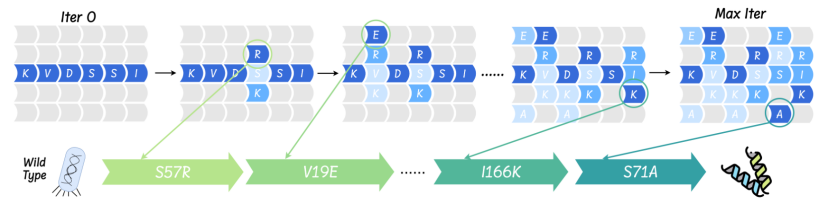
Vérification de l'expérience humide : AMix-1 aide au développement de variantes de protéines AmeR avec une augmentation de 50 fois de l'activité
L'étude a testé la stratégie de « conception contextuelle » dans des expériences en milieu humide, validant ainsi les avantages d'AMix-1 pour la conception efficace de variants AmeR hautement actifs. Les chercheurs ont sélectionné la protéine cible AmeR et utilisé le modèle AMix pour générer 40 variants selon la distribution de probabilité de la famille AmeR. La capacité inhibitrice de chaque variant a été évaluée par des expériences de gènes rapporteurs fluorescents. Chaque variant ne contenait que ≤ 10 mutations d'acides aminés, et plus la valeur de répression était élevée, plus la fonction était forte. De plus, l'étude a proposé un algorithme de mise à l'échelle pour les tests évolutifs afin d'améliorer l'applicabilité d'AMix-1 à l'évolution dirigée des protéines, et a vérifié ses performances grâce à divers indicateurs de zones cibles de simulation informatique.
Les résultats finaux montrent queLa variante optimale générée par AMix-1 présente une amélioration de l'activité jusqu'à 50 fois et ses performances sont améliorées d'environ 77% par rapport au modèle SOTA actuel.De plus, AMix-1 ne repose pas sur un criblage répété ou une conception manuelle, mais est entièrement généré automatiquement par le modèle.Elle a réalisé une boucle fermée complète du « modèle à l'expérience » et a réalisé la toute première avancée dans l'utilisation de la conception de protéines fonctionnelles par l'IA.
Topologie globale, la perception ouvre une nouvelle dimension dans la conception des protéines
Actuellement, la recherche sur l'intégration de l'IA et de la conception de protéines est en plein essor. Outre AMix-1, le modèle de diffusion géométrisé TopoDiff, proposé par l'équipe de Gong Haipeng à l'École des sciences de la vie de l'Université Tsinghua et l'équipe de Xu Chunfu à l'Institut des sciences de la vie de Pékin, a également permis des avancées significatives dans la conception de protéines.
Les modèles de diffusion traditionnels tels que RFDiffusion souffrent non seulement d'un biais de couverture lors de la génération de types de replis spécifiques tels que les immunoglobulines, mais manquent également de mesures d'évaluation quantitative de la topologie globale de la protéine. Cette étude, basée sur des bases de données structurales telles que CATH et SCOPe, a proposé un système non supervisé, le framework TopoDiff. En apprenant et en exploitant des représentations latentes globales conscientes de la géométrie, il permet une génération de protéines inconditionnelle et contrôlable basée sur des modèles de diffusion. Cette étude propose une nouvelle mesure d'évaluation, « Coverage », qui, grâce à un modèle codeur-diffusion en deux étapes, découple la structure protéique en un plan géométrique global et une génération de coordonnées atomiques locales, surmontant ainsi les défis de recherche liés à la couverture des replis protéiques.
De plus, NVIDIA, en collaboration avec Mila, l'Institut québécois d'intelligence artificielle (IQA), a relevé le défi de la prédiction des longues chaînes grâce à un modèle amélioré de génération tout-atome basé sur l'architecture AlphaFold. Les méthodes traditionnelles peinent non seulement à générer des structures tout-atome de très longues chaînes (> 500 résidus), mais ne parviennent pas non plus à explorer les conformations de repliement non classiques, telles que les poches spécifiques aux protéines membranaires. L'équipe de recherche a introduit un mécanisme de prise de décision probabiliste, remplaçant les trajectoires de repliement déterministes par un échantillonnage intégral de chemin issu de la théorie quantique des champs, augmentant ainsi le taux de réussite de la conception des protéines membranaires à 68%.
De la détection géométrique du repliement des protéines à la conception de longues chaînes de plus de 500 résidus, en passant par la conception de protéines pilotée par le langage naturel et le ciblage d'IDP « non médicamentables », l'IA repousse les limites de la conception de protéines et ouvre un nouveau paradigme à la recherche dans ce domaine. À l'avenir, la conception de protéines pilotée par l'IA devrait ouvrir des perspectives encore plus vastes pour le développement de thérapies, d'enzymes et de biomatériaux innovants.
Liens de référence :