Command Palette
Search for a command to run...
L'équipe De David Baker Propose Une Nouvelle Approche Pour Concevoir Des Protéines De Liaison À Des Régions Désordonnées, Ciblant Spécifiquement Des Cibles Non Médicamenteuses, Dans Science.

La plupart des maladies étant directement liées à une fonction anormale des protéines, ces dernières jouent un rôle essentiel dans le développement de médicaments. Lors de la mise au point de nouveaux médicaments, les chercheurs utilisent souvent les protéines comme cibles thérapeutiques principales, permettant ainsi aux médicaments de se lier à des protéines partiellement stables pour intervenir dans le processus pathologique.Cependant, cibler les médicaments sur des protéines intrinsèquement désordonnées (IDP) qui manquent de préférences structurelles, séquentielles et conformationnelles bien définies reste un défi.
La méthode traditionnelle de ciblage par anticorps repose principalement sur la capacité de liaison hautement spécifique des anticorps à des protéines spécifiques, permettant ainsi la reconnaissance et la régulation de ces protéines. Cependant, cette voie de ciblage nécessite non seulement de nombreuses opérations expérimentales, mais les antigènes désordonnés sont également facilement dégradés et inefficaces après injection. Par conséquent,Les protéines avec des régions intrinsèquement désordonnées (IDR) représentant plus de 50% dans le protéome sont généralement considérées comme des cibles « non médicamenteuses » et n'ont jamais été utilisées pour le développement de médicaments.
Dans ce contexte, David Baker, un biologiste computationnel exceptionnel qui a remporté le prix Nobel de chimie 2024 et directeur de l’Institute for Protein Design de l’Université de Washington, et son équipe ont proposé une stratégie de conception de protéines appelée Logos.Sur la base de la stratégie de liaison induite, des protéines de liaison capables de s'adapter à 39 séquences d'acides aminés désordonnées cibles ont été conçues.Cette étude a généré un squelette protéique spécialisé à répétitions étendues, puis l'a généralisé à l'aide du modèle de diffusion RF. Ce squelette intègre des poches spécifiquement conçues pour les séquences peptidiques répétées, permettant au modèle peptidique cible-liant conçu de reconnaître universellement les régions protéiques désordonnées. Cela signifie que davantage de protéines peuvent servir de cibles pour le développement de nouveaux médicaments, ce qui pourrait accélérer la recherche sur le cancer et la maladie d'Alzheimer.
Les résultats de recherche pertinents ont été publiés dans Science sous le titre « Conception de protéines de liaison à des régions intrinsèquement désordonnées ».
Points saillants de la recherche :
*Établir une bibliothèque de structures de modèles adaptée à la reconnaissance générale pour obtenir une induction de conformation d'adaptation de liaison pour n'importe quelle séquence cible.
* Protéines de liaison conçues pour 18 séquences peptidiques synthétiques et 21 régions naturellement désordonnées (IDR) avec une grande diversité et un potentiel thérapeutique, capables de cibler les régions désordonnées des récepteurs extracellulaires associés au cancer et de piloter la localisation des protéines dans les cellules.

Adresse du document :
https://www.science.org/doi/10.1126/science.adr8063
Suivez le compte officiel et répondez « Protéine désordonnée naturelle » pour obtenir le PDF complet
Autres articles sur les frontières de l'IA :
Génération de bibliothèques de modèles : identification universelle des peptides
Cette étude combine des méthodes de conception physique et d'apprentissage profond pour résoudre le problème de liaison des IDR. Limitée par l'incompatibilité des unités peptidiques avec des séquences cibles hétérogènes,L'étude a commencé avec différentes structures de protéines répétées, en utilisant le modèle de diffusion pour réorganiser les poches de liaison des acides aminés dans différentes unités de répétition et les différencier en différents acides aminés et modèles conformationnels.Cela permet une reconnaissance plus large des séquences.
Pour identifier les peptides présents dans les protéines naturellement désordonnées, l'étude a d'abord créé une bibliothèque de modèles de base. Cette bibliothèque présente deux caractéristiques :
*Chaque structure modèle doit être capable d'« envelopper » la conformation de la chaîne peptidique étirée et de fournir un grand nombre d'opportunités d'interactions telles que la liaison hydrogène et l'emballage serré, permettant ainsi une reconnaissance hautement spécifique de la séquence cible.
* La structure du modèle est large et peut correspondre à n'importe quelle séquence cible, de sorte qu'au moins un modèle peut l'induire à devenir une conformation de liaison définie et appropriée.
Le processus de génération d'une bibliothèque de modèles de squelette est divisé en trois étapes : la génération du squelette, la spécialisation des poches actives des protéines et l'assemblage des poches actives des protéines.
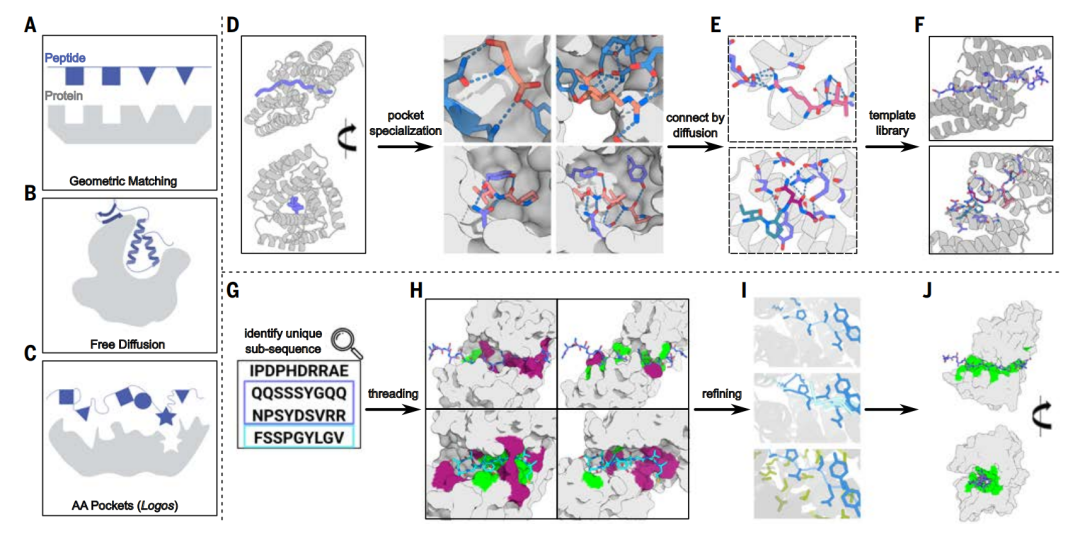
Génération d'échafaudages
Au cours de l'étape de génération de la chaîne principale, l'équipe de recherche a choisi de cibler plusieurs conformations étendues plutôt que de se limiter à la conformation polyproline II, car la conformation polyproline II se produit principalement dans les peptides riches en proline.
Dans la conformation étirée, les chaînes latérales des acides aminés sont alternativement orientées dans des directions opposées, ce qui est cohérent avec les caractéristiques d'une répétition à deux résidus.Les chercheurs ont utilisé la méthode de conception Rosetta pour concevoir une série de séquences répétées de dipeptidiques.Y compris LK, RT, YD, PV et GA (tous sont des abréviations d'acides aminés à une seule lettre), ils sont conçus pour s'enchevêtrer et se lier à ces segments peptidiques dans différentes conformations étirées, de sorte que chaque unité répétitive interagit avec une unité dipeptidique.
Par la suite, les chercheurs ont caractérisé ces versions à quatre unités répétées des protéines de liaison par des expériences de polarisation de fluorescence. Les résultats ont montré une capacité de liaison nanomolaire pour les peptides répétés LK et PV, mais une capacité de liaison plus faible pour les peptides RT et YD, plus polaires, et aucun signal de liaison n'a été détecté pour le gène GA, très flexible.
Spécialisation de poche
Lors de l’étape de spécification de la poche active de la protéine, les chercheurs ont utilisé la modélisation par diffusion pour affiner la poche afin d’obtenir une correspondance plus précise avec la séquence peptidique cible spécifique.
Pour améliorer l'efficacité de la correspondance des modèles, les chercheurs ont affiné la poche de liaison conçue, augmentant le nombre d'unités répétées en interaction de quatre à cinq tout en améliorant la correspondance avec la séquence cible. Cette approche a également renforcé l'affinité entre les structures cibles. Les quatre à neuf acides aminés entourant chaque liaison hydrogène bifurquée de la chaîne latérale entre la protéine répétée et le squelette peptidique sont restés fixes, tandis que les interactions hydrophobes entre les protéines de liaison conçues ont été diversifiées.
L'avantage de cette stratégie réside dans le fait que les exigences de configuration géométrique des liaisons hydrogène sont plus strictes. En comparaison, l'empilement hydrophobe non polaire offre une plus grande liberté spatiale. Par conséquent, lors de la conception, il est plus efficace de conserver les liaisons hydrogène directement à l'aide d'un modèle plutôt que de les échantillonner à plusieurs reprises à partir de zéro.
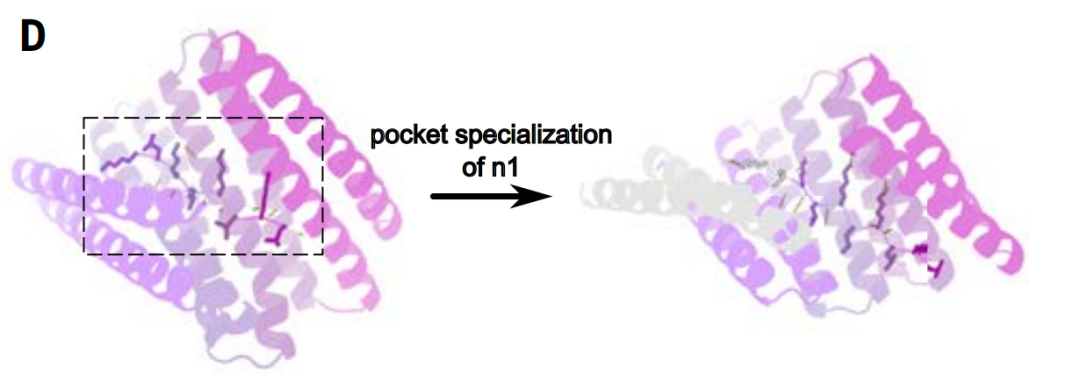
La cinquième structure répétée nouvellement étendue est représentée en gris clair
Assemblage de poche
Lors de l'étape d'assemblage de la poche,Les chercheurs ont utilisé le modèle RFdiffusion pour créer des interfaces entre les poches, ce qui a donné lieu à une structure rigide globale et a généré un modèle pour assembler les poches de liaison dans une nouvelle colonne vertébrale.Les différentes poches du modèle sont disposées selon différents ordres et géométries pour interagir avec la cible peptidique dans une série de conformations étendues, permettant une reconnaissance plus générale des séquences non répétitives.
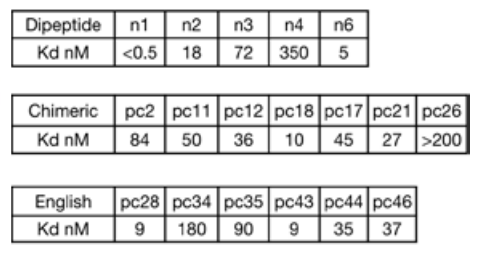
Après avoir généré des modèles de protéines chimériques interagissant avec des cibles peptidiques chimériques, l'étude a localisé paramétriquement les poches de liaison et les a connectées par diffusion radiofréquence. Grâce à cette approche, l'étude a généré 70 propositions de conception pour sept cibles chimériques. La caractérisation par abondance de luciférase fractionnée et par interférométrie en biocouche a révélé qu'avec une moyenne de seulement 10 conceptions testées par cible, six des sept cibles ont atteint une liaison nanomolaire à deux chiffres.
Pour étendre la taille de la bibliothèque de modèles afin de couvrir une plus large gamme de séquences, l'étude a utilisé la technologie d'assemblage de poches pour construire 36 squelettes chimériques contenant des poches qui reconnaissent les résidus polaires, et a généré 1 000 modèles constitués d'une protéine de liaison conçue et d'un squelette peptidique correspondant, dans lequel les acides aminés dans la conformation peptidique peuvent correspondre aux poches conçues dans la protéine de liaison.
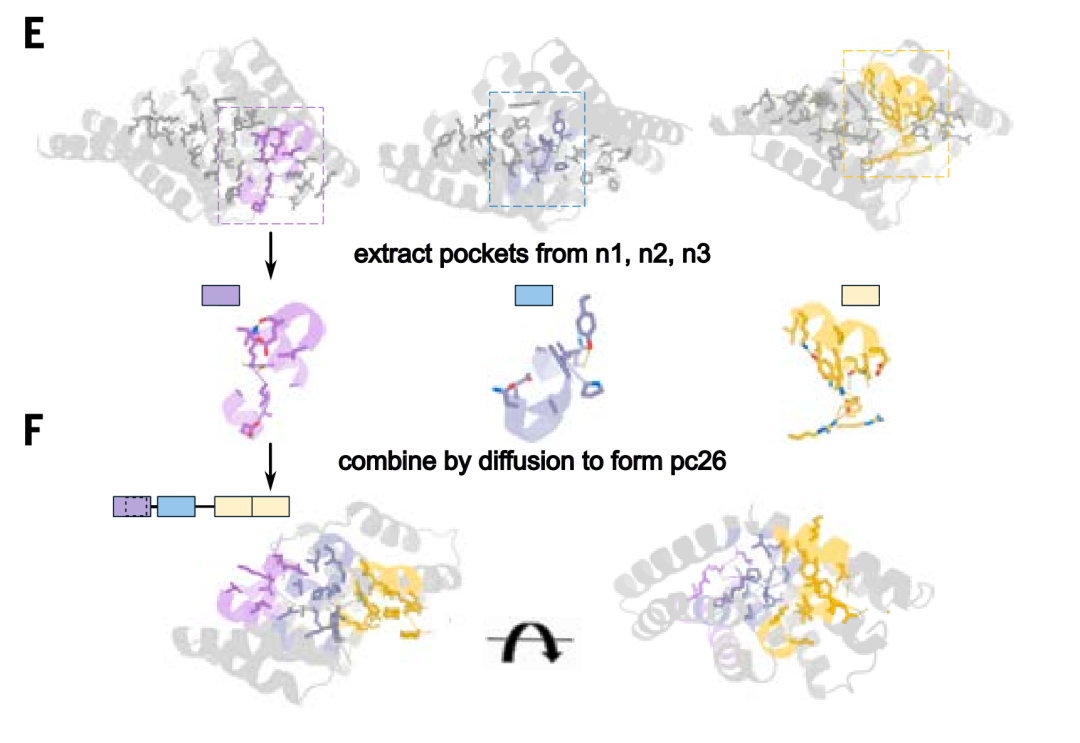
Conception et optimisation des protéines de liaison IDR
Après avoir établi une bibliothèque de modèles, les chercheurs y ont inséré des régions naturellement désordonnées et l'ont utilisée pour générer des protéines de liaison capables de se lier à des séquences synthétiques non répétitives et à toute cible naturellement non structurée. Cette étape se divise en deux parties : la correspondance des threads et le raffinement structurel.
Correspondance de threads : déterminer la paire fragment-modèle de séquence la plus compatible
Dans la correspondance de threads, la séquence cible est intégrée à l'épine dorsale de chaque modèle pour identifier le fragment de séquence le plus compatible à associer au modèle.
D'une manière générale, l'IDP ou l'IDR possède un grand nombre de peptides cibles. Pour identifier le peptide présentant le plus grand potentiel de ciblage dans l'IDR, il faut :L’étude a d’abord éliminé les peptides ayant une faible complexité de séquence et les peptides ayant plusieurs correspondances proches dans le protéome pour éviter les réactions croisées avec les liants de ces cibles.Après un rééchantillonnage local du squelette en mappant les fragments de séquence uniques des acides aminés restants sur le squelette cible de la bibliothèque de modèles,L'étude a utilisé l'outil de conception de séquences protéiques basé sur l'apprentissage profond ProteinMPNN pour optimiser la séquence de la protéine de liaison et l'a évaluée en fonction de l'adéquation entre la protéine de liaison conçue et la séquence cible et de la cohérence entre la valeur de prédiction AF2 et le modèle.
Dans les cas où les mesures AF2 étaient sous-optimales, la RFdiffusion a été utilisée pour personnaliser la structure pour une cible spécifique. La correspondance par thread a ensuite été utilisée pour générer des liants pour les IDP, IDR et fragments d'IDP thérapeutiquement pertinents, générant en moyenne 28 modèles par cible.
Optimisation structurelle : améliorer la correspondance entre la protéine de liaison et le peptide cible
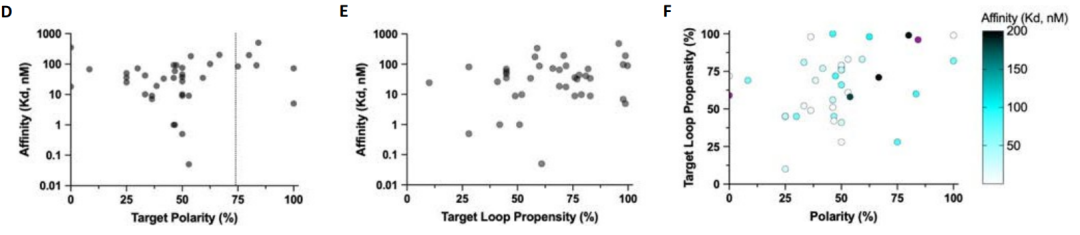
La meilleure correspondance a également été optimisée pour améliorer l’adéquation entre la protéine de liaison conçue et le peptide cible.L'étude a sélectionné la constante de dissociation de la protéine de liaison DYNA_1b1 et de la dynorphine pour les tests, et a optimisé la diffusion par radiofréquence pour le taux de réussite le plus élevé des cibles synthétiques.Les résultats ont montré que parmi les 48 modèles, 45 ont montré une forte affinité dans le test de criblage, et seules les constantes de dissociation de 6 modèles ont montré une faible affinité.
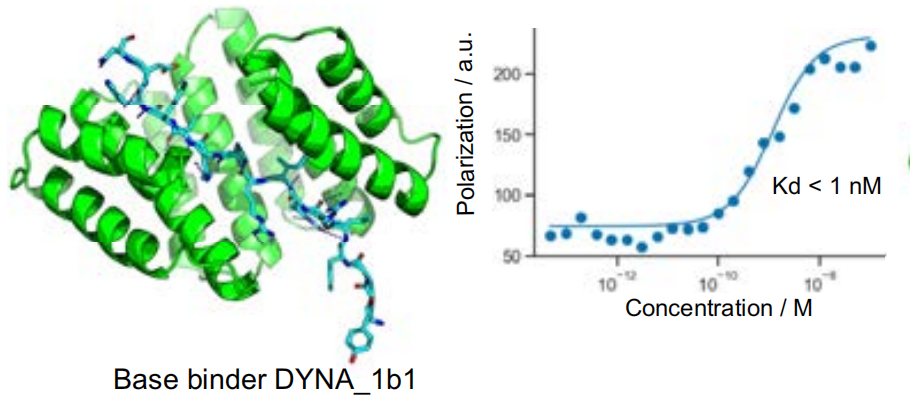
Validation de l'orthogonalité entre la structure de la dynorphine et la protéine de liaison
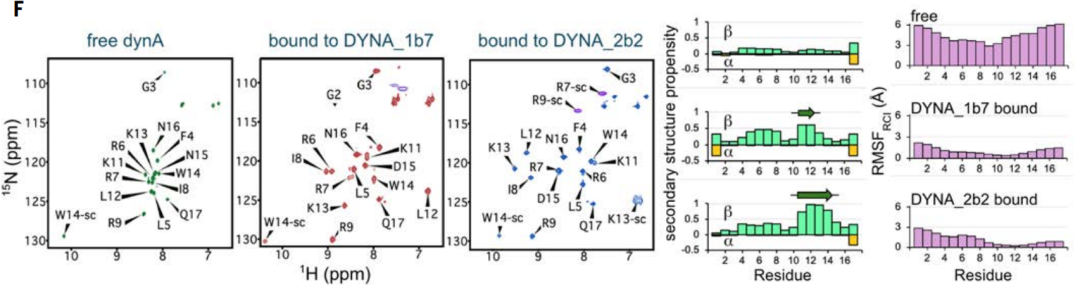
Pour vérifier les changements dans la structure de la dynorphine pendant la liaison, les spectres de résonance magnétique nucléaire (RMN) de la dynorphine A marquée par un isotope ont été détectés en solution lorsqu'elle était non liée, liée à DYNA_1b1 et liée à DYNA_2b2 avec une affinité plus élevée.
D'après les résultats de la RMN, il a été démontré que la dynorphine A libre est intrinsèquement désordonnée, mais les régions contenues dans le squelette conçu deviennent ordonnées lors de la liaison.Pour les deux complexes liés, les données RMN ont révélé des conformations d'état lié étendues, cohérentes avec le modèle conçu et confirmant l'efficacité de l'action de la dynorphine dans l'induction de protéines et de peptides désordonnés dans des conformations non natives.
Pour explorer le potentiel d'optimisation de Logos, les chercheurs ont sélectionné un liant de dynorphine, DYNA_1b1, avec une constante de liaison (Kd) d'environ 1 nM. Ils ont optimisé les modèles les mieux classés par diffusion RF. Sur les 48 modèles, 45 ont démontré une forte liaison à une concentration de 5 nM par criblage BLI, six présentant des valeurs de Kd ≤ 100 pM mesurées par BLI. Les mesures de polarisation de fluorescence de deux des modèles optimisés (DYNA_2b1 et DYNA_2b2) ont révélé des valeurs de Kd inférieures respectivement à 60 pM et 100 pM, comme illustré dans la figure B ci-dessous.
Remarque : la dynorphine est un ligand peptidique du récepteur kappa-opioïde (KOR) associé à la douleur chronique.
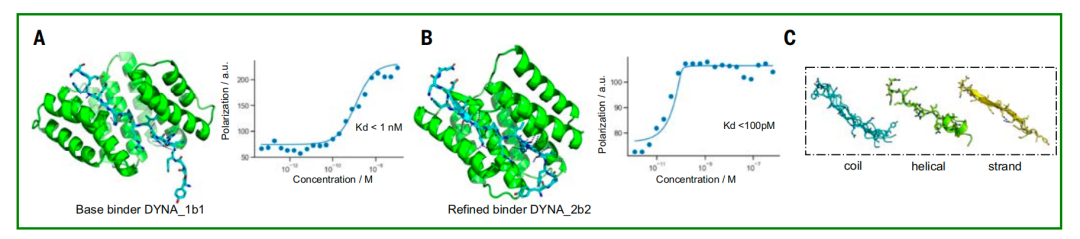
Dans les modèles originaux et optimisés de la dynorphine A, le peptide présentait de multiples conformations, notamment des enroulements aléatoires, des structures partielles en brin β et des structures partielles en hélice α, comme illustré dans la figure C ci-dessus. Bien que les dynorphines A et B partagent une similarité de séquence 62%, leurs protéines de liaison respectives ne se croisent pas et se lient uniquement à leurs cibles respectives. Parallèlement, la structure cocristalline de la protéine DYNA_1b7 conçue et liée à la dynorphine A est parfaitement cohérente avec le modèle de conception computationnelle, notamment au niveau de l'interface de liaison centrale (figures DE ci-dessus). Les données de RMN ont également confirmé queLe squelette de la dynorphine A initialement désordonné devient ordonné après la liaison à la protéine conçue, confirmant une fois de plus l'efficacité du mécanisme d'ajustement induit (Figure F ci-dessus).
Vérifier la fonctionnalité et l'orthogonalité des protéines de liaison
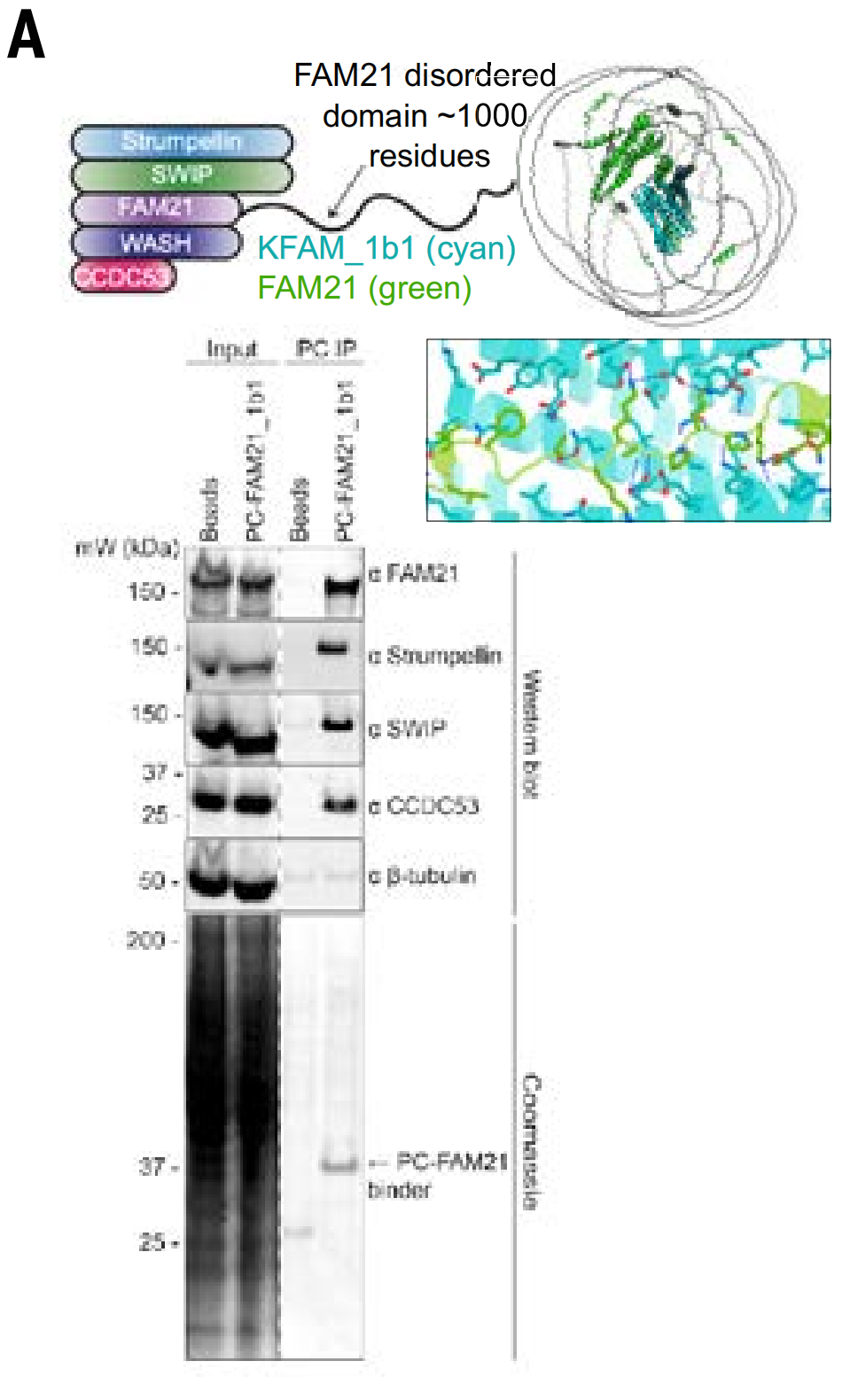
L'étude a utilisé des modèles d'immunoprécipitation des complexes WASH et PER. Le complexe WASH comprend WASH, FAM21, CCDC53, SWIP et WASHC2. Les tests ont montré que FAM21_1b1 extrayait l'intégralité du complexe WASH des lysats cellulaires.
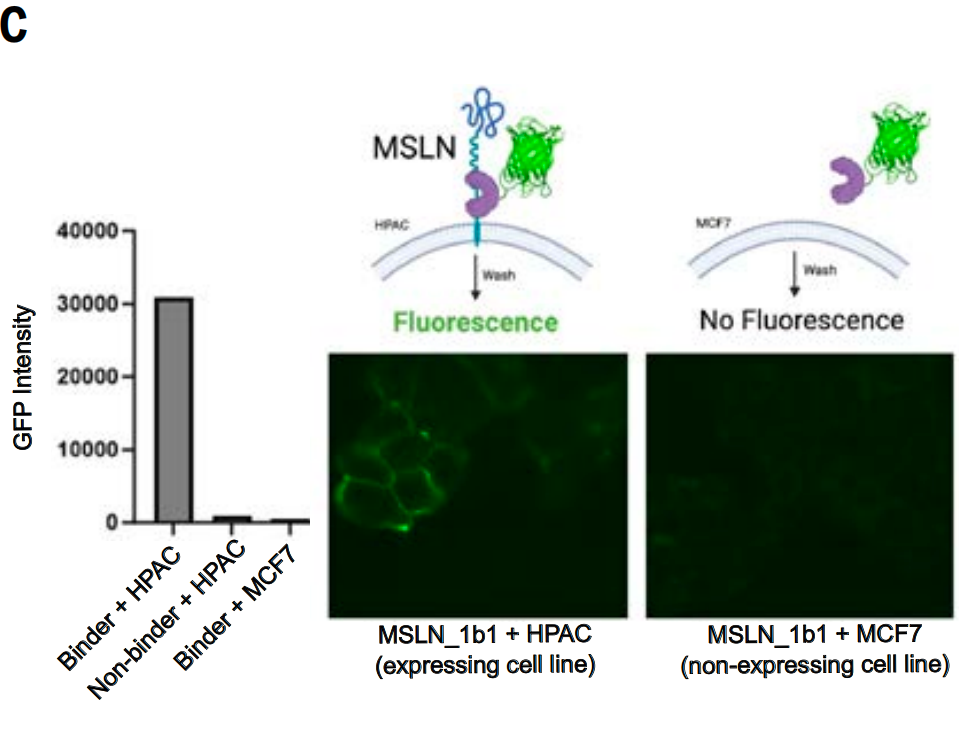
De plus, ils ont également étudié si une protéine de liaison (MSLN_1b1) conçue pour la région juxtamembranaire MSLN peut se lier spécifiquement aux cellules exprimant cette cible (car le clivage de la protéase dans cette région rend la région du domaine extracellulaire plus distale moins adaptée comme cible).
Remarque : la mésothéline (MSLN) est une glycoprotéine de surface cellulaire qui est régulée à la hausse dans de nombreux cancers et qui a donc attiré beaucoup d’attention dans la thérapie ciblée sur les tumeurs.
Les chercheurs ont fusionné la protéine fluorescente verte (GFP) à MSLN_1b1 et l'ont incubée avec des cellules exprimant MSLN (lignée cellulaire d'adénocarcinome pancréatique humain HPAC) et une lignée cellulaire qui n'exprime pas MSLN (lignée cellulaire de cancer du sein MCF7 de la Michigan Cancer Foundation), tout en incluant également une protéine de fusion GFP qui ne se lie pas à MSLN comme contrôle.
La microscopie à fluorescence a révélé que la protéine de fusion GFP-MSLN_1b1 s'agrégait aux jonctions cellulaires des cellules HPAC, ce qui correspond aux caractéristiques de localisation du MSLN. Ce phénomène n'a pas été observé dans les cellules témoins MCF7. De plus, la protéine de liaison témoin n'a montré aucun signal de liaison dans les cellules HPAC, comme le montre la figure C ci-dessous. Par conséquent, MSLN_1b1 reconnaît et se lie spécifiquement au MSLN à la surface cellulaire.
Piloté par l'IA, ouvrant de nouvelles perspectives pour le ciblage des protéines
Actuellement, l'IA est de plus en plus impliquée dans la recherche ciblée sur les protéines, propulsant la recherche vers une nouvelle phase de « parallélisme multi-techniques ». Outre l'équipe de David Baker, les équipes de George M. Burslem et d'Ophir Shalem de l'Université de Pennsylvanie ont également réalisé des avancées révolutionnaires dans la recherche ciblée sur les protéines. Cette équipe a proposé une technologie d'« édition de protéines », utilisant avec succès un système d'intéine fractionnée pour modifier directement la séquence d'acides aminés des protéines après leur synthèse dans des cellules de mammifères vivantes. C'est la première fois que des acides aminés non standard et des marqueurs chimiques (biotine, fluorophores) sont incorporés avec précision dans des protéines endogènes. Les résultats de cette recherche ont été publiés dans la revue Science sous le titre « L'édition intracellulaire de protéines permet l'incorporation de résidus non canoniques dans les protéines endogènes ».
Adresse du document :
https://www.science.org/doi/10.1126/science.adr5499
Par ailleurs, une équipe chinoise et internationale dirigée par Gao Caixia de l'Institut de génétique et de biologie du développement de l'Académie chinoise des sciences et Li Guotian de l'Université agricole de Huazhong, s'appuyant sur les travaux de conception de protéines de l'équipe de David Baker, a développé AiCE, une méthode universelle d'ingénierie des protéines basée sur des modèles de repliement inverse. Grâce à cette stratégie de conception de protéines pilotée par l'IA, ils ont optimisé huit classes de protéines, dont les désaminases et les nucléases, et développé un nouvel éditeur de bases. L'article de recherche, intitulé « Advancing protein evolution with inverse folding models integrate structural and evolutionary constraints », a été publié dans la revue Cell.
Adresse du document :
https://www.cell.com/cell/abstract/S0092-8674(25)00680-4
De l'édition de cellules vivantes aux thérapies neuroprotectrices, des innovations en matière de glycosylation à la conception multi-chaînes basée sur l'IA, grâce aux progrès constants de l'IA en biomédecine, des équipes du monde entier utilisent des approches d'une diversité sans précédent pour relever les défis biomédicaux sous-jacents aux protéines naturellement désordonnées. L'exploration par l'équipe de recherche du ciblage de régions naturellement désordonnées ouvrira la voie à de nouvelles approches thérapeutiques pour des maladies telles que le cancer et la maladie d'Alzheimer.
Liens de référence :








