Command Palette
Search for a command to run...
NVIDIA Réalise Une Percée Dans La Conception De Protéines Au Niveau Atomique, En Générant Des Protéines Contenant jusqu'à 800 Résidus Avec Une Grande Précision
Il est bien connu que la conception de nouvelles protéines dotées de structures et de fonctions spécifiques présente un fort potentiel d'application dans de nombreux domaines tels que le développement de médicaments et la bio-ingénierie. Cependant, cet objectif n'est pas aisé à atteindre, notamment pour ce qui est de comprendre la relation entre la séquence et la structure des protéines, ce qui a toujours constitué un défi majeur lors de la conception de protéines à partir de zéro.
Dans le passé, la plupart des méthodes avaient tendance à concevoir la séquence et la structure des protéines séparément.Par exemple, il faut d'abord générer la séquence, puis la replier, ou concevoir le squelette, puis déterminer la séquence. Cependant, modéliser avec précision la distribution conjointe de la séquence protéique et de la structure atomique reste un défi majeur, afin de maîtriser finement les sites fonctionnels et de réaliser des tâches clés de conception protéique, comme la conception d'échafaudages à motifs atomiques. Cela nécessite non seulement de gérer des séquences discrètes et des coordonnées continues, mais aussi de prendre en compte le problème de l'évolution des dimensions des chaînes latérales avec la séquence.
Dans ce contexte,L'équipe de recherche de NVIDIA et Mila, un institut québécois d'intelligence artificielle au Canada, ont proposé La-Proteina.Il s'agit d'une méthode de conception de protéines au niveau atomique, basée sur l'appariement de flux de potentiel partiel. Elle combine efficacement la modélisation explicite du squelette et la représentation potentielle à taille fixe de chaque résidu pour capturer les informations de séquence et de chaîne latérale atomique, résolvant ainsi le défi majeur de la variabilité dimensionnelle de la représentation explicite des chaînes latérales lors de la génération de protéines et apportant de nouvelles avancées dans le domaine de la conception de protéines.
Les résultats de recherche pertinents ont été publiés sur arXiv sous le titre « La-Proteina : Atomistic Protein Generation via Partially Latent Flow Matching ».
Points saillants de la recherche :
* Un cadre de correspondance de flux partiellement implicite, La-Proteina, est proposé. Il est conçu pour la génération conjointe de séquences protéiques et de structures atomiques complètes. Il combine efficacement une modélisation explicite de la chaîne principale et une représentation implicite de taille fixe de chaque résidu pour capturer à la fois la séquence et les chaînes latérales atomiques.
* Dans des expériences de référence approfondies, La-Proteina atteint des performances SOTA dans la génération de protéines inconditionnelles, capables de générer des protéines à l'échelle atomique diverses, co-conçues et structurellement valides allant jusqu'à 800 résidus.
* L'étude a appliqué avec succès La-Proteina à la conception d'échafaudages de motifs au niveau atomique indexés et non indexés, qui sont deux tâches importantes de conception de protéines conditionnelles, et toutes deux ont démontré que le modèle est supérieur aux précédents générateurs entièrement atomiques.

Adresse du document :
Autres articles sur les frontières de l'IA :
https://go.hyper.ai/owxf6
Ensemble de données : utilisé pour former des modèles inconditionnels, ainsi que des caractéristiques et des fonctions de données protéiques
Cette étude a utilisé 2 ensembles de données pour former des modèles inconditionnels :
L'un d'eux est l'ensemble de données AFDB regroupé par Foldseek, qui est dérivé du filtrage et du regroupement de la base de données AlphaFold (AFDB).Le clustering combine des informations de séquence et de structure, avec un ensemble initial d'environ 3 millions d'échantillons uniques, optimisés selon plusieurs critères : un score pLDDT moyen d'au moins 80, une longueur de protéine comprise entre 32 et 512 résidus, un rapport d'enroulement inférieur à 50% et un maximum de 20 résidus d'enroulement consécutifs. La présence de replis β était spécifiquement requise pour corriger la faible teneur en replis β des protéines générées par le modèle.Finalement, environ 550 000 échantillons de protéines ont été obtenus.Cet ensemble de données a été soigneusement examiné pour rendre les protéines générées par le modèle plus équilibrées dans leurs caractéristiques structurelles, en améliorant notamment le contenu en β-fold.
Le deuxième est un sous-ensemble AFDB personnalisé pour la formation en séquence longue.Les chercheurs ont examiné des échantillons de l’AFDB avec un pLDDT moyen d’au moins 70 et une longueur comprise entre 384 et 896.Après le regroupement, plus de 4 millions de clusters ont été obtenus pour la formation.En se concentrant sur des échantillons de protéines plus longs, il répond aux besoins d'entraînement en séquence longue.
De plus, les données protéiques contiennent des informations sur la séquence (20 types de résidus) et la structure 3D, stockées uniformément grâce à la représentation Atom37. Cette représentation définit un sur-ensemble standardisé de 37 atomes potentiels pour chaque résidu. Une structure protéique de L résidus peut être stockée sous forme de tenseur de forme [L, 37, 3], et les sous-ensembles de coordonnées pertinents sont sélectionnés en fonction du type de chaque résidu.
Cette méthode de standardisation se caractérise par une méthode unifiée de stockage et de représentation des informations structurales des différents résidus, ce qui permet au modèle de traiter uniformément ces informations. Les données à grande échelle d'AFDB fournissent des échantillons riches au modèle, ce qui lui permet d'apprendre un plus large éventail de séquences protéiques et de caractéristiques structurales, améliorant ainsi ses performances et ses capacités de généralisation. Grâce à l'entraînement et à l'expérimentation avec ces données, les modèles associés peuvent mieux saisir la relation entre séquence protéique et structure et obtenir une conception plus précise.
La-Proteina : Architecture innovante et mécanisme de formation du modèle de conception de protéines au niveau atomique
La-Proteina est un modèle innovant pour la conception de protéines à l'échelle atomique. Son principe de base repose sur la « représentation implicite partielle » et vise à résoudre les défis complexes liés à la génération de structures entièrement atomiques.
Au niveau de la conception, compte tenu du défi de générer des structures entièrement atomiques tout en tenant compte du squelette à grande échelle, des types d'acides aminés et des chaînes latérales (les dimensions des chaînes latérales varient selon les acides aminés),La-Proteina propose d'encoder les détails au niveau atomique et le type de résidu de chaque résidu dans un espace latent continu de longueur fixe tout en maintenant une modélisation explicite de l'épine dorsale via les coordonnées α-carbone.
Cette conception présente de nombreux avantages : elle évite non seulement la difficulté de la modélisation mixte par classification continue dans la composante de génération principale du modèle, permettant ainsi à la méthode d'appariement en flux continu complet de générer efficacement des variables cachées, mais elle peut également s'appuyer sur les avancées de la modélisation haute performance de la chaîne principale. Parallèlement, la modélisation explicite de la chaîne principale permet de définir différents calendriers de génération pour le squelette α-carboné global et les détails atomiques des résidus, ce qui est essentiel pour des performances élevées et améliore l'évolutivité, permettant l'extension du modèle aux protéines de grande taille, jusqu'à 800 résidus. Cette approche hybride est la principale raison de sa supériorité par rapport au cadre de modélisation entièrement implicite.
À partir de la structure de composition, comme le montre la figure ci-dessous,Le cœur de La-Proteina est constitué de trois réseaux neuronaux : encodeur, décodeur et débruiteur.Tous les trois partagent l’architecture principale de Transformer basée sur le mécanisme d’attention biaisée.
Parmi eux, l'encodeur est responsable de la cartographie de la protéine d'entrée (contenant des informations de séquence et de structure) aux variables latentes. Sa représentation de séquence initiale couvre les coordonnées atomiques d'origine, les angles de torsion de la chaîne latérale et du squelette et les types de résidus, et la représentation de paire initiale comprend la séparation relative des séquences, la distance par paire et la direction relative entre les résidus ; le décodeur est responsable de la reconstruction de la protéine complète à partir des variables latentes et des coordonnées de l'atome de carbone α, traitant les variables latentes à 8 dimensions et les coordonnées de l'atome de carbone α pour chaque résidu ; le réseau de débruitage est utilisé pour prédire le champ de vitesse qui transfère les échantillons de la distribution de référence gaussienne standard à la distribution de données cible, et conditionne directement le temps d'interpolation dans son bloc Transformer.
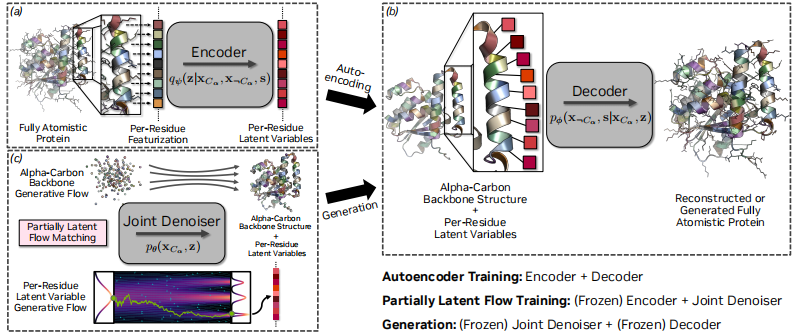
En termes de méthodes de formation,La-Proteina utilise une stratégie d’entraînement en deux phases.
La première étape entraîne un autoencodeur variationnel conditionnel (VAE) : l'encodeur mappe la protéine d'entrée aux variables latentes, et le décodeur reconstruit la protéine à partir de ces variables latentes et des coordonnées atomiques du carbone α. L'ensemble du VAE est optimisé en maximisant la borne inférieure de preuve pondérée β (ELBO). Pour les choix de modélisation ci-dessus, le terme de reconstruction peut être simplifié par la perte d'entropie croisée de la séquence et la perte L2 au carré de la structure.
La deuxième étape optimise le modèle d'appariement de flux pour approximer la distribution cible. Le réseau de débruitage est entraîné en minimisant l'objectif d'appariement de flux conditionnel (CFM). L'utilisation de deux temps d'interpolation distincts, tx et tz, constitue la clé de voûte de cette étape. Ce paramètre permet d'utiliser différents programmes d'intégration pour les coordonnées atomiques du carbone α et les variables latentes lors de l'inférence, améliorant ainsi les performances du modèle.
Grâce à une telle conception et à une telle formation, La-Proteina est capable d'apprendre efficacement la distribution conjointe des séquences protéiques et des structures entièrement atomiques, fournissant ainsi un support technique solide pour la conception de protéines au niveau atomique.
Résultats expérimentaux : La-Proteina est largement en tête dans les quatre tests
Pour vérifier les performances de La-Proteina, l'équipe de recherche a mené une série d'expériences autour de deux axes principaux : la génération inconditionnelle de protéines au niveau atomique et la conception d'échafaudages à motifs atomiques, en considérant de manière exhaustive les performances du modèle dans différents scénarios.
Dans l'expérience de production inconditionnelle de protéines à l'échelle atomique,Comme le montre la figure ci-dessous, l'équipe de recherche a comparé deux variantes de La-Proteina (avec et sans couches de multiplication triangulaires) à l'aide de plusieurs méthodes de référence de génération tout-atome disponibles publiquement, telles que P (tout-atome), APM et PLAID. Les indicateurs d'évaluation couvraient les capacités de conception collaborative tout-atome, la diversité, la nouveauté et les capacités de conception standard.
Les résultats montrent que les deux variantes de La-Proteina surpassent toutes les méthodes de base en termes de capacité de co-conception tout-atome, de capacité de conception et de diversité, et sont également très compétitives en termes de nouveauté.
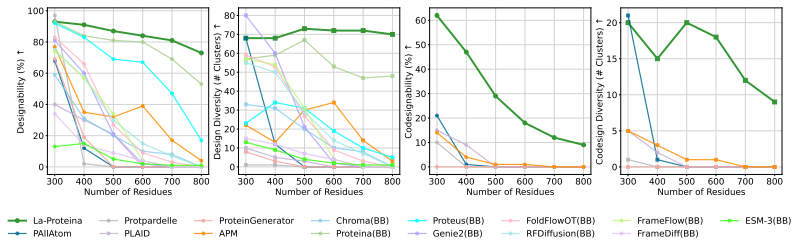
La capacité de La-Proteina à générer de longues chaînes inconditionnelles
Il convient de noter que La-Proteina, qui n'utilise pas de couches de multiplication triangulaires, atteint des performances de pointe tout en ayant une évolutivité élevée, tandis que le deuxième P le plus performant (tout-atome) ne peut traiter que des protéines courtes en raison de sa dépendance à des couches de mise à jour triangulaires coûteuses en termes de calcul.
aussi,L'équipe de recherche a également démontré l'évolutivité de La-Proteina dans la génération de grandes structures entièrement atomiques.Formé sur l'ensemble de données AFDB contenant environ 46 millions d'échantillons, le modèle est plus performant dans la tâche de génération de protéines d'une longueur de plus de 500 résidus, tandis que d'autres méthodes de base entièrement atomiques ont souvent du mal à générer des échantillons efficaces dans cette gamme de longueur.
Dans l’analyse biophysique, l’outil MolProbity a été utilisé pour évaluer la validité du construit.Les résultats ont montré que la structure générée par La-Proteina était de meilleure qualité.Le score est significativement meilleur que toutes les méthodes de base, et la structure générée est plus réaliste au niveau physique et plus similaire à la protéine réelle ; en même temps, en visualisant la distribution des angles dièdres de la chaîne latérale et en les comparant aux références PDB et AFDB, on constate queLa-Proteina peut simuler avec précision l'espace conformationnel des isomères rotationnels des acides aminés,Les méthodes de base s'écartent souvent de la référence, manquant de motifs ou remplissant des zones d'angle irréalistes.
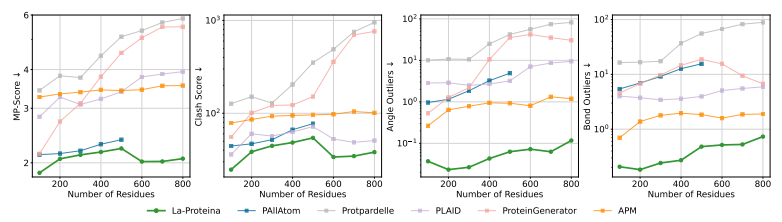
La-Proteina est meilleur que la génération de base tout-atome existante
A une validité de construction plus élevée
Dans l'expérience de conception d'échafaudage à motifs atomiques,L'équipe de recherche a évalué les performances du modèle lors de la tâche de conception d'échafaudages à motifs atomiques, qui exige que le modèle génère une structure protéique capable de supporter avec précision le motif, à partir de la structure atomique d'un motif prédéfini. Les expériences ont été menées dans quatre contextes d'évaluation, dont la conception d'échafaudages atomiques tout-atomique et avancée, ainsi que des versions indexées et non indexées.
Les résultats montrent que dans les quatre contextes,La-Proteina surpasse considérablement la seule méthode de base comparable sur tous les atomes, Protpardelle, et est capable de résoudre avec succès la plupart des tâches de référence.En particulier pour les motifs constitués de 3 segments résiduels différents ou plus, la version non indexée de La-Proteina fonctionne mieux que la version indexée, probablement parce que la fixation des positions de plusieurs segments limite la flexibilité du modèle pour explorer différentes solutions structurelles.
Avancées scientifiques et pratiques innovantes dans le domaine de la conception de protéines au niveau atomique
Dans le domaine de la conception des protéines, l'orientation de recherche sur la conception des protéines au niveau atomique représentée par La-Proteina a suscité un vif intérêt auprès du monde universitaire et du monde des affaires. De nombreuses universités et entreprises ont réalisé d'importantes avancées scientifiques et adopté des pratiques innovantes dans ce domaine.
Dans le milieu universitaire, certaines équipes de recherche s'efforcent d'améliorer les performances et l'évolutivité des modèles de génération de protéines. Par exemple, NVIDIA s'est associée à Mila, à l'Institut québécois d'intelligence artificielle, à l'Université de Montréal et au Massachusetts Institute of Technology (MIT) pour développer un modèle de génération de protéines.Le Proteina développé a été formé sur la base de données AlphaFold à grande échelle (AFDB).A démontré l'évolutivité d'un modèle basé sur le flux pour la génération de structures protéiques.
Certaines études utilisent également des modèles de diffusion pour la conception de protéines. Par exemple, les premiers générateurs de protéines basés sur la diffusion, tels que RFDiffusion et Chroma, se concentrent sur la génération de squelettes. Des études ultérieures ont élargi le champ d'application des modèles de diffusion pour la conception de protéines, notamment la diffusion sur des variétés SO(3) et les méthodes d'appariement de flux euclidien.
Certaines équipes de recherche se concentrent également sur la modélisation conjointe de la séquence et de la structure des protéines. Par exemple, ProtComposer, lancé par NVIDIA et le MIT, utilise des modèles statistiques auxiliaires et des primitives 3D pour générer des structures protéiques. D'autres travaux traitent des structures entièrement atomiques en modélisant conjointement le squelette et la séquence des protéines ou en utilisant des modèles à variables latentes. De plus, des modèles de langage ont également été appliqués à la conception des protéines : certaines méthodes se concentrent sur les séquences protéiques, tandis que d'autres tokenisent les informations structurelles et modélisent conjointement séquences et structures.
Dans le monde des affaires, Cradle, une société de biotechnologie néerlandaise, se concentre sur l'utilisation de l'intelligence artificielle pour simplifier le processus de conception des protéines. Elle a mis en place un laboratoire humide pour accumuler des milliards de séquences et de données protéiques afin d'entraîner des modèles d'intelligence artificielle générative propriétaires, simplifiant ainsi la conception et l'optimisation des protéines. Xaira Therapeutics, un fournisseur américain de services pharmaceutiques en IA, s'engage à créer des molécules adaptatives pour des indications spécifiques grâce à ses atouts en matière de recherche avancée en apprentissage automatique, de génération de données à grande échelle et de développement thérapeutique. Certaines entreprises s'engagent également à combiner la technologie de conception des protéines avec l'intelligence artificielle et l'apprentissage automatique afin d'améliorer l'efficacité et la précision de la conception des protéines.
Les avancées scientifiques de ces universités et les pratiques innovantes des entreprises ont apporté une riche expérience et un soutien technique au développement de la conception des protéines, favorisant ainsi le développement continu de ce domaine. Grâce aux progrès technologiques constants, la conception des protéines devrait jouer un rôle important dans de nombreux domaines à l'avenir.
Articles de référence :
1.https://mp.weixin.qq.com/s/7r69S3XpNMjemo3EiXzNeQ
2.https://mp.weixin.qq.com/s/DrZEdsb1SqSSkv_hbrp3TA








