Command Palette
Search for a command to run...
En Traitant Simultanément Les Informations De La Chaîne Principale Et De La Chaîne Latérale Des Protéines, Stanford Et al. Ont Réalisé Une Modélisation De La Structure Atomique Complète Basée Sur Un Réseau Neuronal De Transmission De messages.
La conformation des chaînes latérales des protéines désigne la disposition spatiale spécifique des chaînes latérales des résidus d'acides aminés des protéines dans l'espace tridimensionnel. Son étude peut aider à comprendre la relation entre structure et fonction des protéines et présente une grande valeur applicative en ingénierie des protéines, en conception de médicaments et dans d'autres domaines. Cependant, les méthodes actuelles de conception de séquences protéiques basées sur l'apprentissage profond se concentrent principalement sur la conception de séquences protéiques à chaîne principale fixe, et la plupart d'entre elles sont incapables de modéliser la conformation des chaînes latérales des protéines lors de la génération de séquences.Les interactions clés de la chaîne latérale sont déduites uniquement sur la base de la géométrie de la chaîne principale et des étiquettes de séquence d'acides aminés connues, tout en ignorant le rôle de la conformation de la chaîne latérale des protéines dans les protéines.
Pour combler cette lacune, une équipe de l’Université de Stanford et de l’Arc Institute de Palo Alto, en Californie,Ensemble, nous avons proposé une nouvelle méthode de conception de séquences protéiques, FAMPNN (Full-Atom MPNN), qui peut modéliser explicitement l'identité de séquence et la structure de la chaîne latérale de chaque résidu d'acide aminé.Le modèle utilise une architecture de transmission de messages basée sur des réseaux de neurones graphes (GNN), combinée à des modules MPNN (Message Passing Neural Networks) et GVP (Geometric Vector Perceptron) améliorés pour le codage tout-atomique, et peut traiter simultanément les informations des chaînes principales et latérales des protéines. Des études ont montré que le FAMPNN peut améliorer significativement la qualité de la conception des séquences protéiques et la précision des prédictions expérimentales en modélisant explicitement les structures tout-atomiques.
Le résultat de la recherche, intitulé « Conditionnement et modélisation de la chaîne latérale pour la conception de séquences protéiques à atomes complets avec FAMPNN », a été sélectionné pour l'ICML 2025.
Points saillants de la recherche :
* Nous introduisons une méthode qui combine les objectifs d'entropie croisée et de perte de diffusion pour modéliser la distribution par étiquette de l'identité de séquence discrète des résidus et de la structure continue de la chaîne latérale.
* Nous mettons en œuvre une méthode d'échantillonnage itérative légère pour générer des échantillons à partir d'une distribution conjointe et utilisons des couches MPNN et GVP améliorées pour le codage tout-atome
* Des recherches ont montré que FAMPNN peut améliorer efficacement la précision de la conception des séquences et la prédiction expérimentale de la fitness des protéines en modélisant explicitement les structures entièrement atomiques.

Adresse du document :
Autres articles sur les frontières de l'IA :
https://go.hyper.ai/owxf6
Ensembles de données : divers ensembles de données optimisent la formation et l'évaluation des modèles
Pour garantir l'efficacité et la fiabilité du modèle, l'équipe de recherche a utilisé des ensembles de données multiples complexes pour la formation et l'évaluation :
L’étude a principalement utilisé l’ensemble de données S40 de CATH 4.2.L'ensemble de données est un ensemble organisé de domaines extraits de la Protein Data Bank (PDB), avec des domaines redondants avec une homologie dépassant 40% supprimés, et divisé en ensembles d'entraînement, de validation et de test dans un rapport de 8:1:1.
L'ensemble de données PDB est construit sur la base de l'ensemble de la base de données PDB et comprend des structures publiées au 30 septembre 2021. Les chercheurs ont regroupé les protéines en fonction de l'homologie de séquence de 40% au niveau de la chaîne protéique et ont priorisé les exemples de protéines multi-chaînes pour les modèles de formation afin d'apprendre à concevoir des protéines multi-chaînes.
Les ensembles de données CASP13, 14 et 15 sont principalement utilisés pour évaluer les performances du modèle dans le conditionnement de la chaîne latérale.L'équipe de recherche a utilisé la recherche de suggestions MMseqs2 pour supprimer toutes les séquences présentant une homologie supérieure à 40% avec les ensembles de données CASP13, 14 et 15 des ensembles de données de formation et de validation, puis a mesuré les performances d'emballage de la chaîne latérale par l'écart quadratique moyen (RMSD) entre les chaînes latérales prédites et les véritables chaînes latérales.
L’ensemble de données SKEMPlv2 a été utilisé pour évaluer la capacité prédictive du modèle pour l’affinité de liaison protéine-protéine.L'ensemble de données rassemble les affinités de liaison mesurées expérimentalement de milliers de variantes de séquences dans des centaines d'interactions protéine-protéine, et après traitement, l'ensemble de données final comprend 6 649 points de données.
Les ensembles de données S669, Megascale et FireProtDB ont été utilisés pour évaluer la capacité de prédiction zéro coup du modèle pour la stabilité des protéines.Ces ensembles de données contiennent des mesures expérimentales des variations de stabilité de diverses protéines naturelles (△△G), qui sont des ensembles de données de référence largement utilisés pour les prédicteurs de stabilité. L'ensemble de données Megascale est une version sélectionnée et dédupliquée de l'ensemble de données. L'équipe de recherche a fusionné l'ensemble d'apprentissage, l'ensemble de validation et l'ensemble de test en un seul ensemble de données, obtenant ainsi un ensemble de données contenant 272 712 points de données expérimentaux impliquant 298 protéines différentes. L'ensemble de données FireProtDB contient les variations d'énergie libre de 3 438 mutations uniques de 100 protéines uniques, dont 3 420 exemples ont finalement été utilisés après traitement. L'ensemble de données S669 contient des mesures expérimentales de 669 mutations uniques de 94 protéines, et quatre variants ont été exclus de l'ensemble de données en raison de la présence d'acides aminés non standard.
Les ensembles de données CR9114, CR6261 et G6 ont été utilisés pour évaluer les performances du modèle dans la prédiction de l'affinité de liaison anticorps-antigène.Parmi eux, l'ensemble de données CR9114 contient toutes les combinaisons possibles de 16 substitutions d'acides aminés. L'ensemble de données CR6261 contient toutes les combinaisons possibles de 11 substitutions d'acides aminés, avec un total respectif de 65 536 et 2 048 séquences. L'ensemble de données G6 contient un total de 4 275 points de données se liant au VEGF-A.
Outil intelligent pour comprendre simultanément la séquence des protéines et la structure de la chaîne latérale
L'objectif principal de cette étude est de permettre au modèle d'apprendre simultanément la séquence protéique et la conformation de la chaîne latérale. À cette fin, l'équipe de recherche a utilisé la modélisation du langage masqué pour entraîner FAMPNN en se basant sur la cohérence des séquences.La formation est réalisée de manière de bout en bout, en combinant la perte d'entropie croisée catégorielle (pour la prédiction de séquence) et la perte de diffusion (pour la prédiction de conformation de chaîne latérale).Cela permet au modèle de restaurer simultanément la séquence masquée et ses conformations de chaîne latérale correspondantes sur la base de séquences partiellement connues et de coordonnées de chaîne latérale.
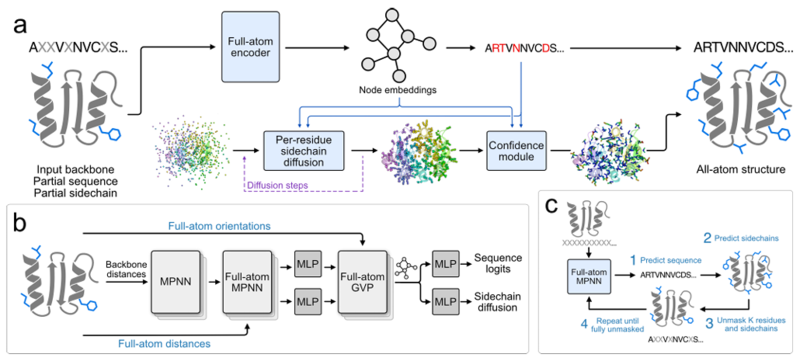
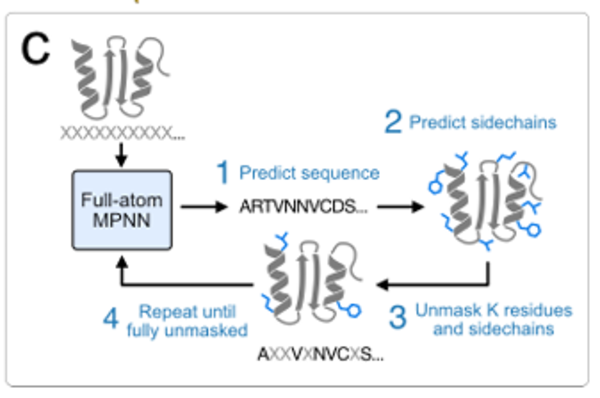
En termes d'échantillonnage, une stratégie d'échantillonnage itérative similaire à celle de MaskGIT est mise en place. Elle part de l'état où la séquence et la chaîne latérale sont complètement masquées, puis prédit et supprime progressivement le masque de certaines séquences et jetons de chaîne latérale jusqu'à obtenir la structure complète de la séquence et de la chaîne latérale. Comme illustré dans la figure ci-dessous :
Dans la conception spécifique, les coordonnées de la chaîne latérale sont représentées au format atom37.Chaque résidu est une matrice de taille fixe 37 x 3 contenant 37 atomes, incluant les coordonnées 3D de 4 atomes de la chaîne principale (N, Cα, C et O) et 33 atomes de la chaîne latérale. Pour les chaînes latérales sans type d'atome spécifique, des atomes fantômes (positionnés à la position Cα du résidu) sont utilisés pour les représenter. Cette méthode résout le problème des différences de nombre d'atomes de la chaîne latérale selon les acides aminés.
En tant que cœur de l'extraction de fonctionnalités, l'encodeur tout atome utilise un réseau neuronal graphique d'architecture hybride MPNN-GVP pour l'encodage.L'architecture se compose de trois composants principaux : l'encodeur invariant de la chaîne principale, l'encodeur invariant tous atomes et l'encodeur équivariant tous atomes. Les deux premiers composants s'appuient sur l'architecture de ProteinMPNN. L'encodeur invariant de la chaîne principale est identique à l'encodeur MPNN, qui code uniquement la structure de la chaîne principale ; l'encodeur invariant tous atomes remplace le décodeur MPNN, identique à l'encodeur MPNN de la chaîne principale, mais dont la caractérisation est étendue à tous les atomes. Le dernier composant utilise un GVP amélioré pour permettre au modèle de raisonner sur les orientations interatomiques vectorielles en plus des distances interatomiques scalaires précédemment codées.
En termes de génération de coordonnées de chaîne latérale, l'équipe de recherche a adopté la méthode de diffusion euclidienne par jeton.Le cœur de cette méthode repose sur l'utilisation du modèle de diffusion euclidien (EDM) pour résoudre le problème de la génération de valeurs continues des coordonnées atomiques des chaînes latérales. L'objectif est de générer une structure de chaîne latérale qui corresponde à la structure de la chaîne principale et à la disposition spatiale des acides aminés environnants. Lors de l'apprentissage, un bruit aléatoire est d'abord ajouté aux coordonnées réelles des chaînes latérales, puis le modèle est autorisé à supprimer ce bruit et à restaurer les coordonnées réelles en fonction du niveau de bruit et des informations connues. Lors de l'inférence, à partir des coordonnées du bruit aléatoire, le modèle supprime progressivement le bruit et génère des coordonnées de chaîne latérale proches des coordonnées réelles.
Dans le même temps, afin d'éviter l'influence de la rotation et de la translation globales de la protéine sur la génération de la chaîne latérale, les coordonnées atomiques de la chaîne latérale sont converties en un système de coordonnées local basé sur les atomes de la chaîne principale pendant la formation, puis reconverties en système de coordonnées global après la génération.L'entrée du modèle de diffusion comprend les caractéristiques extraites par l'encodeur tout atome, l'identité de séquence prédite et le niveau de bruit actuel.Les coordonnées de la chaîne latérale générées sont également utilisées dans la fonction de perte conjointe pour guider la formation du modèle, comme illustré dans la figure ci-dessous.
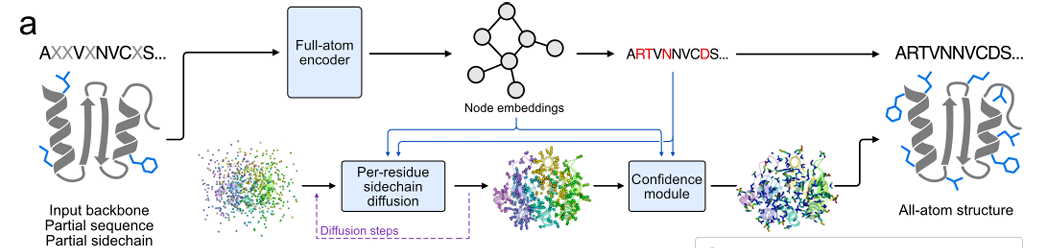
Afin de réduire l'erreur de prédiction du modèle et d'améliorer la précision, l'équipe de recherche a également conçu un module de confiance pour prédire l'erreur de compression de la chaîne latérale (erreur de chaîne latérale prédite, pSCE). Plus précisément,Ce module divise l'erreur réelle des atomes de la chaîne latérale (la distance entre les coordonnées générées et les vraies coordonnées) en 33 intervalles.Le modèle est entraîné avec une perte d'entropie croisée catégorielle, ce qui lui permet de prédire l'intervalle de chaque erreur atomique à partir des informations du processus de génération, puis d'obtenir l'estimation d'erreur finale pSCE grâce à l'espérance de la probabilité d'intervalle. Les données d'entrée de ce module incluent les caractéristiques du codeur tout-atome, la séquence générée et les coordonnées de la chaîne latérale, ainsi que le niveau de bruit du processus de diffusion. La sortie pSCE reflète efficacement la précision de l'empilement de la chaîne latérale, ce qui est utile pour analyser des résultats de conception de haute qualité et améliorer l'interprétabilité du modèle, améliorant ainsi l'évaluation de la qualité de la génération de la structure de la chaîne latérale.
Résultats expérimentaux : Les performances sont nettement meilleures que le modèle basé uniquement sur la chaîne principale
Afin de vérifier les performances et d'évaluer précisément le modèle, l'équipe de recherche a d'abord mené des expériences de récupération de séquence et d'évaluation de l'autocohérence, puis comparé FAMPNN à d'autres méthodes. Les objets de comparaison spécifiques sont présentés dans la figure ci-dessous.
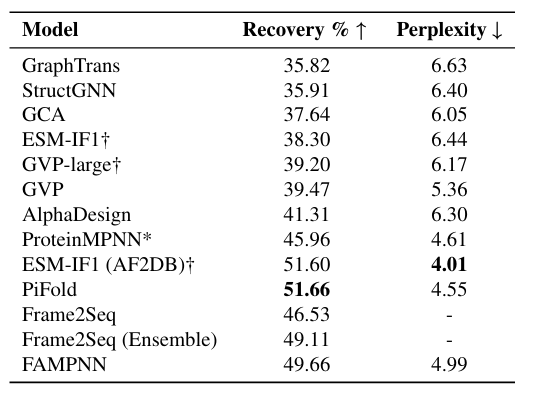
Les expériences montrent queFAMPNN surpasse les méthodes de pointe actuelles en termes de précision de récupération de séquence en une seule étape, atteignant 49,66%.En comparaison, ProteinMPNN est seulement de 45,96% et GVP est seulement de 39,47%.
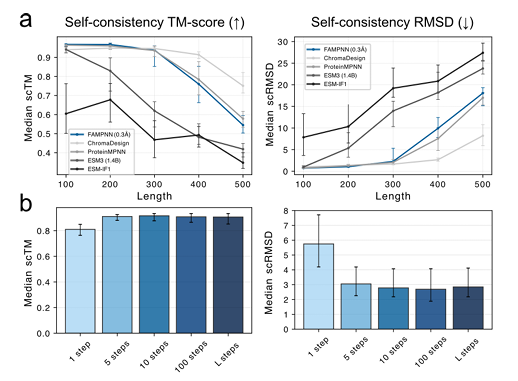
Dans l'évaluation d'auto-cohérence sur la nouvelle chaîne principale générée sur la base de RFdiffusion,FAMPNN (0,3 Å) est comparable à ProteinMPNN en termes de mesures scTM (similarité structurelle) et scRMSD (écart quadratique moyen).Dix étapes d'échantillonnage itératif permettent d'atteindre un degré élevé d'autocohérence, plus efficace que la méthode autorégressive complète. Comme le montre la figure suivante :
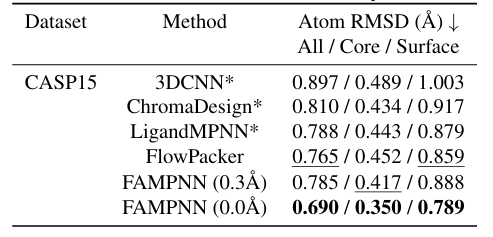
En termes d'emballage de chaîne latérale, les chercheurs ont comparé le modèle proposé avec d'autres méthodes sur les ensembles de données CASP13, 14 et 15.Les expériences montrent que dans l'évaluation de la structure cristalline de l'ensemble de tests CASP15, le RMSD atomique (Tout/Cœur/Surface) de FAMPNN (0,0 Å) est de 0,690/0,350/0,789 Å, ce qui est meilleur que les autres méthodes.Il existe une forte corrélation avec l'erreur de chaque atome et celle de chaque résidu, avec des coefficients de corrélation de Spearman de 0,843 et 0,780 respectivement. Comme le montre la figure ci-dessous :
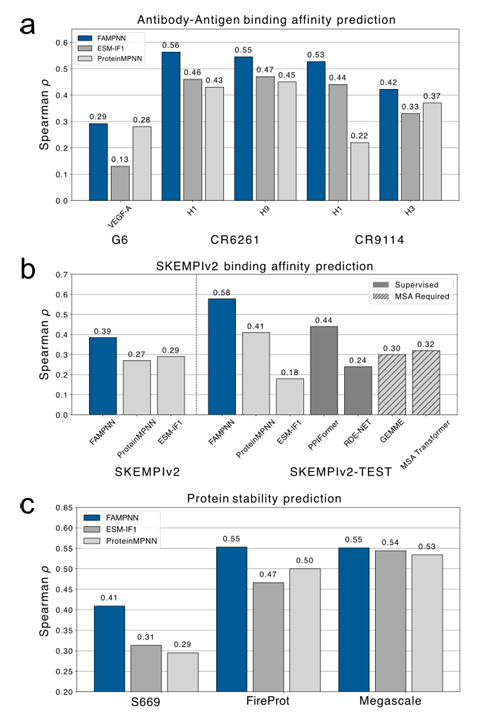
Dans l'évaluation de l'aptitude des protéines dans des conditions entièrement atomiques,Sur l'ensemble de données SKEMPlv2, FAMPNN a nettement surpassé le modèle non supervisé, et même le modèle supervisé sur le sous-ensemble test, démontrant une forte capacité de généralisation dans la prédiction à échantillon zéro. Sur les trois ensembles de données de stabilité S669, Megascale et FireProtDB, FAMPNN a obtenu des résultats légèrement supérieurs à ProteinMPNN et ESM-IF ; pour la prédiction de l'affinité de liaison anticorps-antigène, FAMPNN a toujours surpassé les méthodes non supervisées les plus avancées, ProteinMPNN et ESM-IF1, ce qui démontre l'utilité de FAMPNN pour la stabilité des protéines et l'amélioration des interactions protéine-protéine. Comme le montre la figure suivante :
Dans des expériences évaluant si la modélisation tout-atome peut améliorer les performances de conception de séquences,L’étude a révélé que l’ajout de cibles d’emballage de chaîne latérale et de paramètres de conditions pour tous les atomes peut améliorer la précision de la séquence.De plus, les performances du FAMPNN et du modèle de base s'amélioreront à mesure que davantage d'informations structurelles seront injectées. À l'interface protéine-protéine, la modélisation des interactions des chaînes latérales est plus cruciale, et fournir un contexte partiel des chaînes latérales en conjonction avec la séquence peut améliorer considérablement la précision par rapport à un contexte de séquence partiel.
De plus, comparé au ligand MPNN, le FAMPNN exploite plus efficacement le contexte de la chaîne latérale et effectue un compactage de la chaîne latérale en fonction d'un nombre différent de contextes de séquences partielles ou de conformations de la chaîne latérale. Plus le nombre de contextes est élevé, plus la précision du compactage est élevée. Comme le montre la figure c ci-dessous :
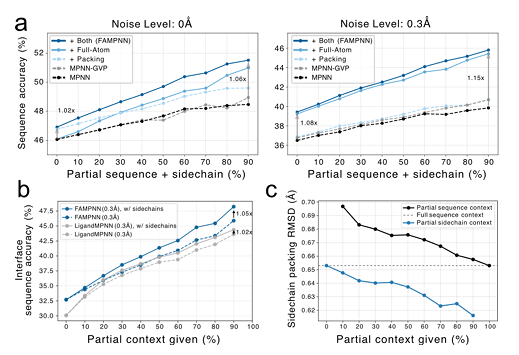
En résumé, les expériences ci-dessus démontrent que FAMPNN présente des avantages significatifs dans la prédiction de la forme physique des protéines par rapport aux modèles basés uniquement sur la chaîne principale.
Porté par l'intelligence artificielle, le monde universitaire est en plein essor dans le domaine de la modélisation des sidechains
Comme mentionné au début, la conformation de la chaîne latérale est cruciale pour la fonction des protéines. Cependant, une fois la chaîne principale de la protéine déterminée, de nombreuses conformations de chaîne latérale sont encore possibles, ce qui rend leur modélisation et leur étude complexes et difficiles à maîtriser. Outre cette étude, de nombreux instituts de recherche universitaires du monde entier font progresser la recherche sur la modélisation des chaînes latérales grâce à des technologies de pointe en apprentissage profond et aux connaissances biologiques.
Une équipe de l’Université Fudan en Chine a proposé une méthode de modélisation de chaîne latérale en deux étapes appelée OPUS-Rota5.Cette méthode utilise un 3D-Unet amélioré pour capturer les caractéristiques de l’environnement local.Les informations sur les ligands de chaque résidu sont incluses, puis le module RotaFormer est utilisé pour agréger différents types de caractéristiques. Des évaluations sur des ensembles de tests incluant CAMEO et CASP15 montrent qu'OPUS-Rota5 surpasse significativement d'autres méthodes de modélisation des chaînes latérales de pointe. L'étude associée a été publiée sur ScienceDirect sous le titre « OPUS-Rota5 : Une méthode de modélisation des chaînes latérales protéiques très précise avec 3D-Unet et RotaFormer ».
Adresse du document :
https://www.sciencedirect.com/science/article/pii/S0969212624001266
Une équipe de l’Université de Pékin a proposé une autre méthode, appelée GeoPacker.Cette méthode combine l’apprentissage profond géométrique avec ResNet pour modéliser les chaînes latérales des protéines. GeoPacker représente explicitement les interactions atomiques de manière invariante en rotation et en translation afin d'extraire des informations de position relative. En termes de précision de prédiction de la structure des chaînes latérales, GeoPacker surpasse les méthodes les plus avancées basées sur les fonctions d'énergie et est environ 10 fois et 700 fois plus rapide que les méthodes d'apprentissage profond DLPacker et OPUS-Rota4, respectivement, avec une précision de prédiction comparable. Les recherches associées ont été publiées sous le titre « GeoPacker : Un nouveau cadre d'apprentissage profond pour la modélisation des chaînes latérales des protéines ».
Adresse du document :
https://onlinelibrary.wiley.com/doi/epdf/10.1002/pro.4484
Dans le même temps, l’équipe de l’Université de Toronto a proposé un modèle appelé FlowPacker.Son objectif est de prédire avec précision la forme spécifique de la chaîne latérale en fonction de la séquence d’acides aminés connue et de la structure de la chaîne principale de la protéine.Comparé aux méthodes avancées précédentes, FlowPacker est plus performant sur la plupart des indicateurs et s'exécute plus rapidement. Par exemple, il présente des avantages en termes d'erreur de prédiction d'angle, de proximité entre l'angle prédit et la valeur réelle, d'écart de position atomique, etc. Les recherches associées ont été publiées sous le titre « FlowPacker : Compactionnement des chaînes latérales des protéines avec adaptation du flux torsionnel ».
Adresse du document :
De manière générale, le décodage des conformations des chaînes latérales est crucial pour le développement des sciences de la vie. Le développement continu des technologies d'intelligence artificielle a incontestablement favorisé l'essor rapide de la biologie structurale et de la biologie computationnelle, et a également permis aux instituts de recherche nationaux et étrangers d'atteindre des résultats académiques florissants. Une fois ces avancées mises en œuvre du laboratoire à l'application, elles provoqueront inévitablement une nouvelle vague de bouleversements dans le domaine des sciences de la vie et propulseront les sciences biologiques et médicales vers un nouveau chapitre.








