Command Palette
Search for a command to run...
Modélisation De Séquence Longue De 8 000, Modèle De Langage Protéique Prot42 Peut Générer Des Liants À Haute Affinité En Utilisant Uniquement La Séquence Protéique Cible
Les liants protéiques (tels que les anticorps et les peptides inhibiteurs) jouent un rôle irremplaçable dans des scénarios clés tels que le diagnostic des maladies, l'analyse d'imagerie et l'administration ciblée de médicaments.Traditionnellement, le développement de liants protéiques hautement spécifiques s’appuie largement sur des techniques expérimentales telles que l’affichage de phages et l’évolution dirigée.Cependant, ces méthodes sont généralement confrontées aux défis d’une consommation énorme de ressources et de longs cycles de R&D, et sont limitées par le goulot d’étranglement inhérent à la complexité des combinaisons de séquences protéiques.
Avec le développement de l'intelligence artificielle, les modèles de langage protéique (MLP) sont devenus un outil essentiel pour comprendre la relation entre séquences et fonctions protéiques. Pour la conception de liants protéiques, les MLP permettent de concevoir directement des protéines ligands ou des fragments d'anticorps présentant une forte affinité de liaison à partir de la séquence protéique cible, grâce à la capacité de génération des modèles de langage. Cependant, cette approche se heurte également à des difficultés, notamment l'absence de MLP dotés à la fois de capacités de modélisation à long contexte et de véritables capacités de génération, notamment pour la conception d'interfaces de liaison complexes et de liants protéiques longs. Il existe un écart technique important.
Sur cette base, une équipe de recherche conjointe de l'Inception AI Institute à Abu Dhabi, aux Émirats arabes unis et de Cerebras Systems dans la Silicon Valley, aux États-Unis,La première famille de PLM, Prot42, qui s'appuie uniquement sur les informations de séquence protéique et ne nécessite pas d'entrée de structure tridimensionnelle, a été proposée.Ce modèle exploite la puissance génératrice de l’autorégression et de l’architecture décodeur uniquement.Permet la génération de liants protéiques à haute affinité et de protéines de liaison à l'ADN spécifiques à la séquence en l'absence d'informations structurelles.Prot42 a obtenu de bons résultats dans les expériences de référence PEER, de génération de liants protéiques et de génération de liants spécifiques à la séquence d'ADN.
La recherche connexe s'intitule « Prot42 : une nouvelle famille de modèles de langage protéique pour la génération de liants protéiques sensibles à la cible » et a été publiée sous forme de pré-impression sur arXiv.
Points saillants de la recherche* Prot42 utilise une stratégie d'entraînement par expansion progressive du contexte, qui s'étend progressivement des 1 024 acides aminés initiaux à 8 192 acides aminés. * Dans le test de référence PEER, Prot42 obtient de bons résultats dans 14 tâches telles que la prédiction de la fonction des protéines, la localisation subcellulaire et la modélisation des interactions. * Contrairement à AlphaProteo, qui s'appuie sur une structure 3D, Prot42 n'a besoin que de la séquence protéique cible pour générer des liants.
Adresse du document :
Autres articles sur les frontières de l'IA :
https://go.hyper.ai/UuE1o
Le projet open source « awesome-ai4s » rassemble plus de 100 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Ensembles de données : 3 grands ensembles de données prennent en charge le développement et la formation de modèles
Dans cette étude, plusieurs ensembles de données clés ont été utilisés pour entraîner et évaluer les performances du modèle. Ces ensembles de données couvrent non seulement un large éventail d'informations sur les séquences protéiques, mais incluent également des données d'interaction protéine-ADN, fournissant ainsi de riches ressources d'entraînement pour Prot42.
Base de données sur les interfaces protéine-ADN (PDIdb) 2010
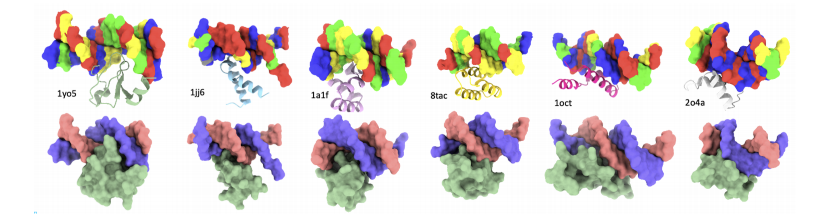
Pour concevoir des protéines capables de se lier à des séquences d’ADN cibles, les chercheurs ont utilisé l’ensemble de données PDIdb 2010.Cet ensemble de données contient 922 paires ADN-protéine uniques et a été utilisé pour former et évaluer la capacité de Prot42 à générer des protéines se liant à des séquences d'ADN spécifiques.Pour évaluer les quatre modèles ADN-protéine, les chercheurs ont extrait des fragments d’ADN de diverses structures PDB, notamment 1TUP, 1BC8, 1YO5, 1L3L, 2O4A, 1OCT, 1A1F et 1JJ6.
Ensemble de données UniRef50
L'ensemble de données de pré-formation du modèle Prot42 provient principalement de la base de données UniRef50.La base de données contient 63,2 millions de séquences d’acides aminés, couvrant un large éventail d’espèces biologiques et de fonctions protéiques.Ces séquences sont regroupées et les séquences présentant une similarité supérieure à 50% sont regroupées, réduisant ainsi la redondance des données et améliorant l'efficacité de la formation.
Avant de former Prot42, l’équipe de recherche a prétraité l’ensemble de données UniRef50.Ils sont étiquetés à l’aide d’un vocabulaire de 20 acides aminés standards.Utilisez Xtoken pour représenter les résidus d'acides aminés (X est utilisé pour marquer les résidus d'acides aminés inhabituels ou ambigus).
Dans la phase de prétraitement des données,L'équipe de recherche a traité les séquences avec une longueur de contexte maximale de 1 024 jetons et a exclu les séquences plus longues que cela, obtenant finalement un ensemble de données filtré de 57,1 millions de séquences.La densité de remplissage initiale est de 27%. Afin d'améliorer l'utilisation des données et l'efficacité de calcul, l'équipe de recherche a adopté une stratégie de remplissage à longueur de séquence variable (VSL).Nous avons maximisé le taux d'occupation des jetons dans une longueur de contexte fixe et avons finalement réduit l'ensemble de données à 16,2 millions de séquences complétées.L'efficacité de remplissage atteint 96%.
base de données STRING
La base de données STRING est une base de données complète sur les interactions protéine-protéine.Il intègre des données expérimentales, des prédictions informatiques et des résultats d'exploration de texte pour fournir des scores de confiance pour les interactions protéiques. Afin d'entraîner Prot42 à générer des liants protéiques, l'équipe de recherche a sélectionné des paires d'interactions protéiques avec des scores de confiance ≥ 90% dans la base de données STRING afin de garantir la haute fiabilité des données d'apprentissage.De plus, la longueur de la séquence a été limitée à 250 acides aminés pour se concentrer sur des protéines de liaison à domaine unique gérables.Après le criblage, l'ensemble de données final contient 74 066 paires d'interactions protéine-protéine, un ensemble d'entraînement D(train)(pb) contenant 59 252 échantillons et un ensemble de validation D(val)(pb) contenant 14 814 échantillons.
Architecture du modèle : 2 variantes majeures dérivées de l'architecture du décodeur autorégressif
Prot42, mentionné dans cet article, est un PLM basé sur une architecture de décodeur autorégressif qui génère des séquences d'acides aminés une par une et prédit l'acide aminé suivant à partir de l'acide aminé précédemment généré. Cette architecture permet au modèle de capturer les dépendances à longue distance dans la séquence.Il est capable d'apprendre des représentations riches directement à partir de grandes bases de données de séquences protéiques non étiquetées, comblant ainsi efficacement le fossé entre le grand nombre de séquences protéiques connues et la proportion relativement faible de séquences protéiques (<0,3%).Dans le même temps, le modèle contient plusieurs couches de transformateurs, chacune contenant un mécanisme d'auto-attention multi-têtes et un réseau neuronal à rétroaction pour capturer des modèles complexes dans la séquence.
Sa conception s'inspire des avancées en matière de traitement du langage naturel, notamment du modèle LLaMA. Prot42 capture les informations évolutives, structurelles et fonctionnelles des protéines en les pré-entraînant sur des séquences protéiques non marquées à grande échelle, permettant ainsi la génération de liants protéiques à haute affinité.
Sur cette base,Les chercheurs ont pré-entraîné 2 variantes de modèles,C'est-à-dire Prot42-B et Prot42-L.
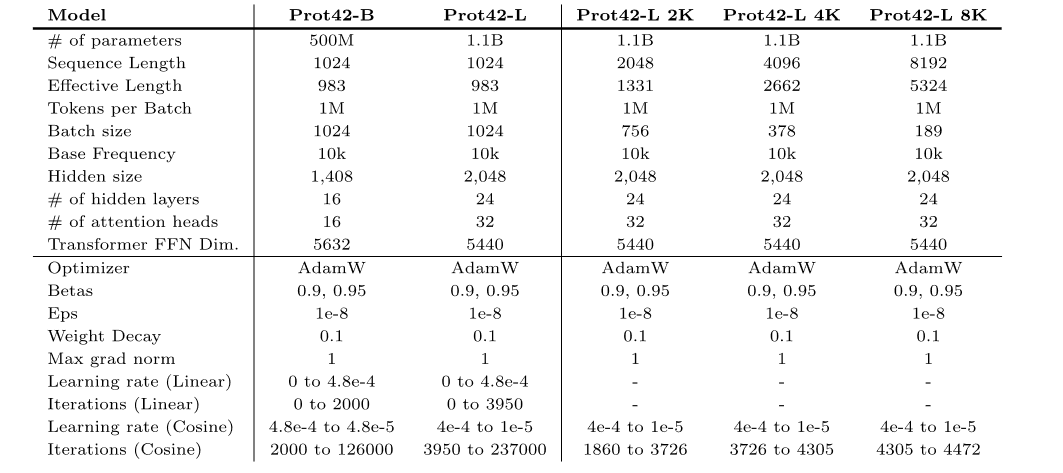
* Prot42-B :Dans la version de base, le modèle dispose de 500 millions de paramètres et prend en charge une longueur de séquence maximale de 1 024 acides aminés.
* Prot42-L :La version étendue dispose de 1,1 milliard de paramètres de modèle et prend également en charge une longueur de séquence maximale de 1 024 acides aminés.La longueur du contexte de Prot42-L a été progressivement étendue de 1 024 acides aminés à 8 192 acides aminés.Dans ce processus, une longueur de contexte progressivement croissante et une taille de lot constante (1 million de jetons non remplis) ont été utilisées pour garantir la stabilité et l'efficacité du modèle lors du traitement de longues séquences, améliorant considérablement la capacité du modèle à traiter de longues séquences et des structures protéiques complexes.Prot42-L contient également 24 couches cachées, chacune avec 32 têtes d'attention.La dimension de la couche cachée est de 2 048.
Conclusion expérimentale : Un grand potentiel est démontré dans les 6 tâches
Pour évaluer les performances du modèle Prot42 avant la validation sur les tâches en aval, les chercheurs ont utilisé la mesure standard de la complexité paramétrique (PPL) pour évaluer les modèles de langage autorégressifs, à savoir les performances du modèle Prot42 à différentes longueurs de contexte.Tous les modèles ont une perplexité relativement élevée à 1 024 jetons, mais s'améliorent considérablement à environ 6,5 à 2 048 jetons.Les résultats montrent que le modèle de base et les modèles optimisés pour des contextes plus courts présentent des performances similaires pour leurs longueurs de contexte maximales respectives. Les performances du modèle de contexte 8 000 sont particulièrement remarquables : bien que sa perplexité soit légèrement supérieure pour les séquences de longueur moyenne (2 048 à 4 096 jetons), il est capable de gérer des séquences jusqu'à 8 192 jetons et atteint une perplexité minimale de 5,1 à la longueur maximale.Après plus de 4 096 jetons, la courbe de perplexité montre une tendance à la baisse.Comme le montre la figure ci-dessous.
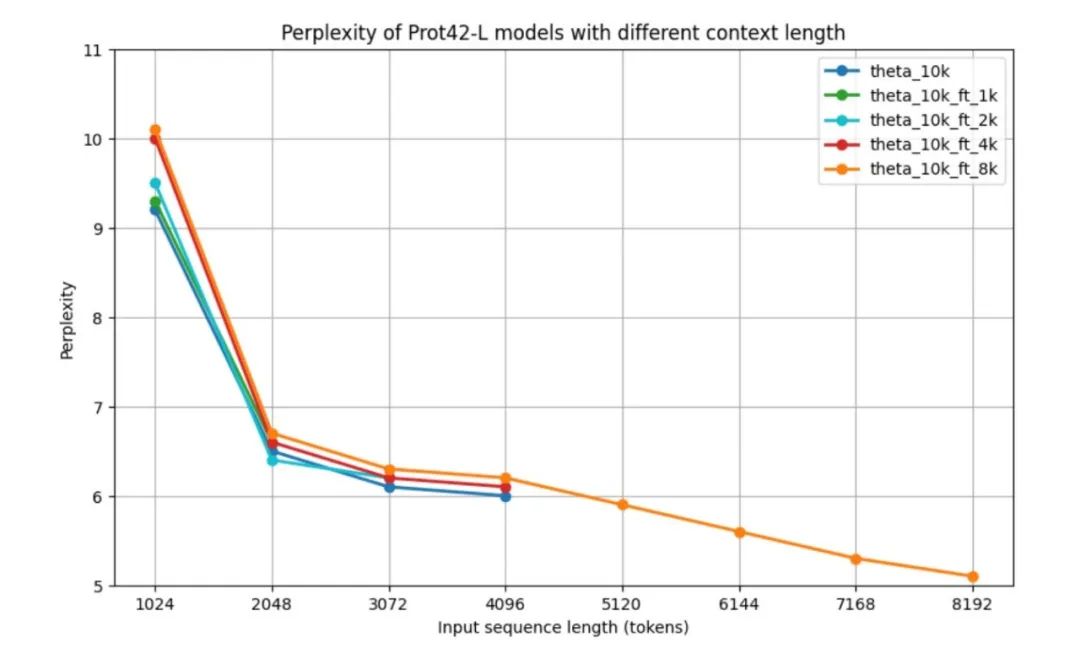
À mesure que la longueur du contexte augmente, le PPL du modèle diminue progressivement, indiquant que la capacité du modèle à traiter de longues séquences a été considérablement améliorée.En particulier, le modèle de contexte 8K atteint le PPL le plus bas, indiquant qu'il peut utiliser efficacement la fenêtre de contexte étendue pour capturer les dépendances à longue portée dans les séquences de protéines.La fenêtre de contexte élargie constitue une avancée majeure dans le domaine de la modélisation des séquences protéiques, permettant une représentation plus précise des protéines complexes et des interactions protéine-protéine, ce qui est essentiel pour générer des liants protéiques efficaces.
Grâce à une série d’évaluations expérimentales rigoureuses,Prot42 a démontré d’excellentes performances dans plusieurs tâches clés.Il a été démontré qu’il est efficace dans la génération de liants protéiques et la conception de protéines se liant à des séquences d’ADN spécifiques.
Prédiction de la fonction des protéines
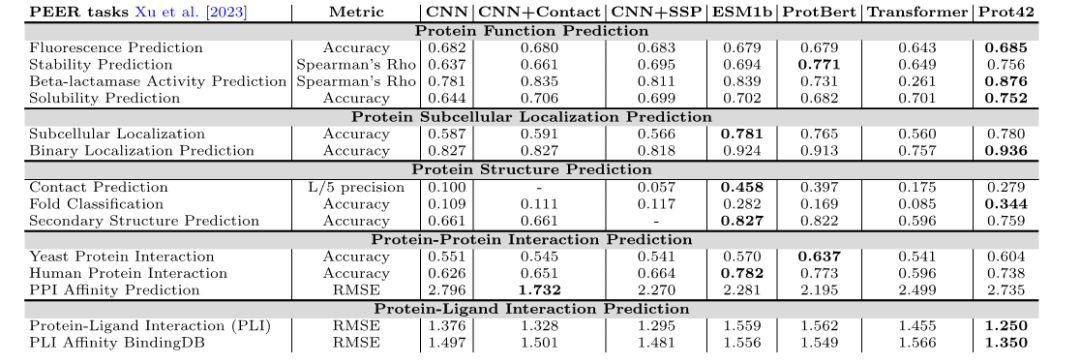
Lors du test de référence PEER, le modèle Prot42 a obtenu de bons résultats dans plusieurs tâches de prédiction de la fonction protéique, notamment la prédiction de la fluorescence, de la stabilité, de l'activité des β-lactamases et de la solubilité. Comparé aux modèles existants,Prot42 a obtenu des avantages significatifs dans la prédiction de la stabilité, la prédiction de la solubilité et la prédiction de l'activité β-lactamase.Cela indique son grand potentiel dans les tâches d’ingénierie des protéines à haute résolution.
Prédiction de la localisation subcellulaire des protéines
Les chercheurs ont représenté chaque séquence protéique sous forme de vecteur de grande dimension de 32 × 2048, ont intégré le modèle Prot42-L dans la séquence protéique entière et ont effectué des calculs. Afin d'évaluer intuitivement la différenciation de la qualité de l'intégration et des compartiments, les chercheurs ont appliqué l'intégration stochastique des voisins distribuée en t (t-SNE) pour réduire la dimensionnalité, rendant ainsi la visualisation des groupes protéiques plus claire.Il a été vérifié que Prot42 fonctionne bien dans la tâche de prédiction de la localisation subcellulaire des protéines, et sa précision est comparable à celle des modèles avancés existants.Grâce à une analyse visuelle, l’équipe de recherche a vérifié davantage l’efficacité du modèle Prot42 dans la capture des caractéristiques de localisation subcellulaire des protéines.
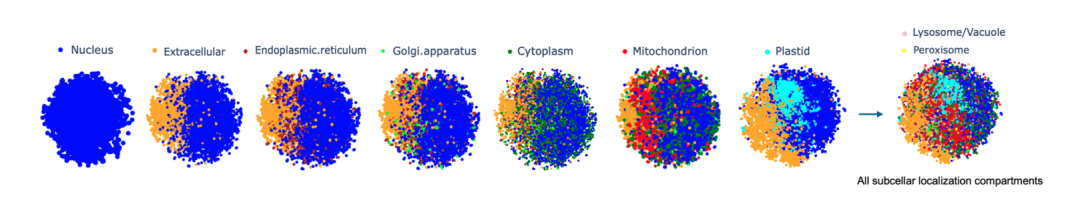
Prédiction de la structure des protéines
Dans la tâche de prédiction de la structure des protéines,Le modèle Prot42 a obtenu d’excellents résultats dans la prédiction des contacts, la classification des pliages et la prédiction des structures secondaires.Ces résultats indiquent que le modèle Prot42 est capable de capturer des différences subtiles dans la structure des protéines, fournissant un support solide pour la modélisation d'interactions biologiques complexes et les applications pharmaceutiques.
Prédiction de l'interaction protéine-protéine
Dans les tâches de prédiction de l'interaction protéine-protéine et de l'interaction protéine-ligand, le modèle Prot42 a montré une grande précision et une grande fiabilité.Les chercheurs ont utilisé Chem42 pour générer des vecteurs d’intégration chimique et les ont comparés à ChemBert., en tant que modèle de représentation chimique, ses indicateurs de performance restent néanmoins supérieurs à ceux des méthodes existantes et proches des résultats obtenus avec Chem42. En particulier, lors de l'utilisation de Chem42 pour générer des inclusions chimiques, ses résultats de prédiction sont proches de ceux des modèles chimiques professionnels.Cela indique que Prot42 a une bonne extensibilité dans la combinaison des informations chimiques.Fournit un soutien solide à la conception de médicaments.
Génération de liants protéiques
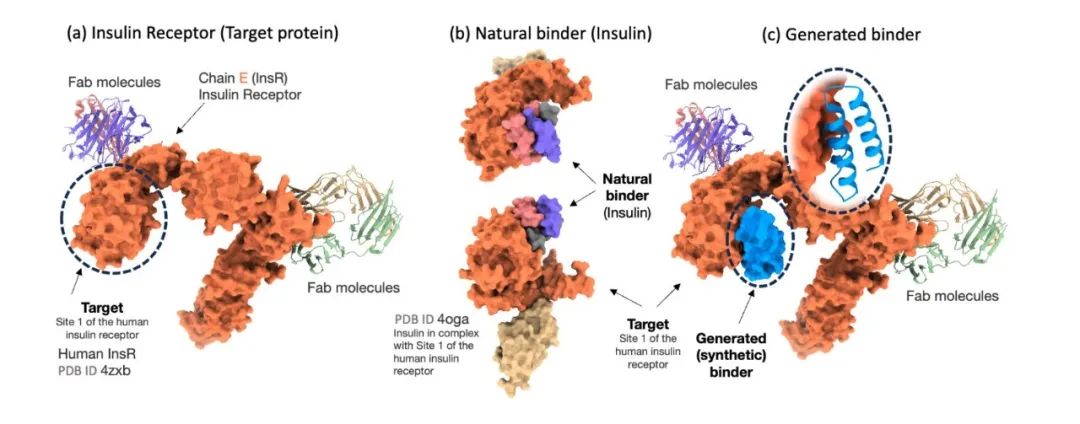
Afin d'évaluer rigoureusement l'efficacité du modèle Prot42 dans la génération de liants protéiques, les chercheurs l'ont comparé à AlphaProteo, un modèle avancé conçu spécifiquement pour la prédiction des liants protéiques. Les résultats expérimentaux ont montré queLe modèle Prot42 a généré des liants avec de fortes affinités prédites sur plusieurs cibles thérapeutiquement pertinentes.En particulier sur des cibles telles que IL-7Rα, PD-L1, TrkA et VEGF-A,Le modèle Prot42 a obtenu des résultats nettement meilleurs que le modèle AlphaProteo.Ces résultats indiquent que le modèle Prot42 présente des avantages significatifs dans la génération de liants protéiques, comme le montre la figure ci-dessous.
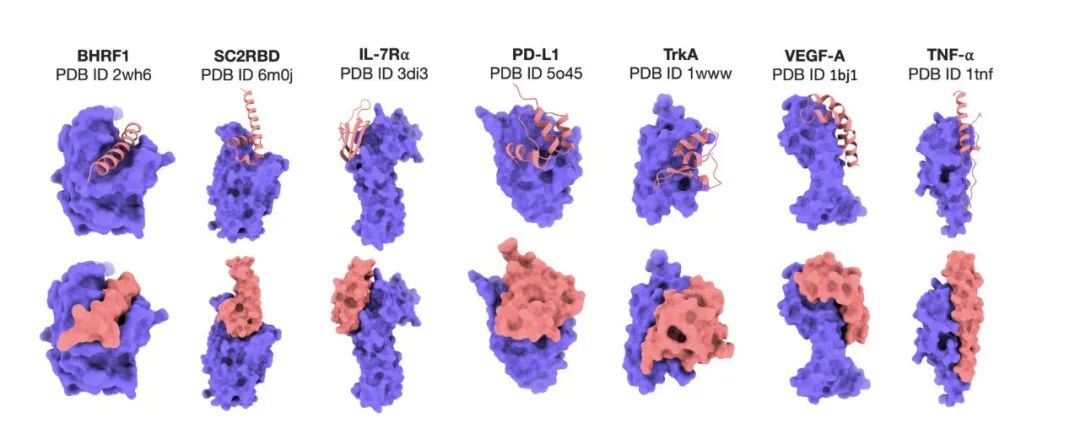
Génération de liants spécifiques à la séquence d'ADN
Lors de l'expérience de génération de liants spécifiques à une séquence d'ADN, Prot42 a également obtenu des résultats remarquables. Les résultats expérimentaux ont montré queEn combinant une stratégie multimodale d'intégration de gènes et d'intégration de protéines, le modèle Prot42 est capable de générer des protéines qui se lient spécifiquement aux séquences d'ADN cibles et présentent une affinité élevée.La spécificité de liaison évaluée par le modèle DeepPBS était élevée. Ces résultats indiquent que le modèle Prot42 présente également un fort potentiel pour la génération de liants spécifiques de séquences d'ADN, offrant ainsi de nouveaux outils pour la régulation des gènes et l'édition génomique.
Percées et innovations en intelligence artificielle dans la conception des protéines
Avec l'intégration profonde des biotechnologies et de l'intelligence artificielle, le domaine d'avenir de la conception des protéines connaît des transformations révolutionnaires. En tant qu'organes essentiels du vivant, l'analyse structurale et fonctionnelle des protéines a toujours constitué un point délicat de la recherche scientifique. L'intervention de l'IA accélère la résolution de ce casse-tête complexe, ouvrant de nouvelles perspectives pour des scénarios tels que le développement de nouveaux médicaments et la transformation par ingénierie enzymatique.
Ces dernières années, la technologie de l’IA a de nouveau fait des percées, et les nouvelles technologies centrées sur l’IA générative poussent la conception des protéines vers le stade de la « Genèse ».
L'équipe du professeur Xu Dong de l'Université du Missouri a proposé le modèle de langage protéique sensible à la structure (S-PLM), qui aligne les informations sur la séquence protéique et la structure 3D dans un espace latent unifié en introduisant l'apprentissage par contraste multi-vues.Nous utilisons Swin Transformer pour traiter les informations structurelles prédites par AlphaFold et les fusionner avec l'intégration de séquences basée sur ESM2 pour créer un PLM sensible à la structure.L'article « S-PLM : Modèle de langage protéique sensible à la structure via un apprentissage contrastif entre séquence et structure » a été publié dans Advanced Science. S-PLM intègre intelligemment les informations structurelles dans la représentation des séquences en alignant les séquences protéiques avec leurs structures tridimensionnelles dans un espace latent unifié. Il explore également des stratégies d'ajustement fin efficaces, permettant au modèle d'atteindre d'excellentes performances dans différentes tâches de prédiction de protéines, marquant une avancée majeure dans le domaine de la prédiction de la structure et de la fonction des protéines.
Adresse du document :
https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202404212
De plus, l'équipe de recherche de l'Université Tsinghua et d'autres chercheurs ont proposé un modèle de langage protéique unifié, xTrimoPGLM. Ce modèle de base et cadre de pré-apprentissage unifié peut être étendu à 100 milliards de paramètres et est conçu pour diverses tâches liées aux protéines, notamment la compréhension et la génération (ou conception). Utilisant le modèle de langage général (GLM) comme pilier de ses objectifs d'attention bidirectionnelle et d'autorégression, ce modèle se distingue des précédents PLM basés uniquement sur l'encodage ou le décodage causal. Cette étude a exploré la compréhension unifiée et le pré-apprentissage de la génération de PLM à très grande échelle, a révélé de nouvelles possibilités pour la conception de séquences protéiques et a favorisé le développement d'un éventail plus large d'applications liées aux protéines. Elle a été publiée dans la sous-revue Nature sous le titre « xTrimoPGLM : transformateur pré-entraîné unifié à 100 milliards de paramètres pour le déchiffrement du langage des protéines ».
Adresse du document :
https://www.nature.com/articles/s41592-025-02636-z
La percée de Prot42 constitue non seulement une avancée technique, mais témoigne également de la maturité progressive du modèle de conception « data-driven + IA » dans le domaine des sciences de la vie. À l'avenir, l'équipe de recherche prévoit de vérifier les liants générés par Prot42 par des expériences et de compléter l'évaluation computationnelle par des tests fonctionnels réels. Cette étape consolidera l'utilité du modèle dans les applications pratiques et améliorera sa précision prédictive, comblant ainsi le fossé entre la génération de séquences par IA et la biotechnologie expérimentale.
Références :
1.https://arxiv.org/abs/2504.04453
2.https://mp.weixin.qq.com/s/SDUsXpAc8mONsQPkUx4cvA
3.https://mp.weixin.qq.com/s/x7_Wnws35Qzf3J0kBapBGQ
4.https://mp.weixin.qq.com/s/SDUsXpAc8mONsQPkUx4cvA








