Command Palette
Search for a command to run...
Le MIT Et Harvard Ont Proposé Conjointement PUPS En Intégrant Un Modèle De Langage Protéique Et Un Modèle d'inpainting d'image Pour Obtenir Une Localisation Protéique Unicellulaire
La localisation subcellulaire d'une protéine fait référence à l'emplacement spécifique d'une protéine dans la structure cellulaire.Ceci est essentiel pour que les protéines puissent remplir leurs fonctions biologiques. Pour donner un exemple simple, si nous imaginons une cellule comme une immense entreprise, dans laquelle le noyau cellulaire, les mitochondries, la membrane cellulaire, etc. correspondent à différents départements tels que le bureau du président, le département de production d'énergie et le gardien, alors ce n'est que lorsque la protéine correspondante entre dans le bon « département » qu'elle peut fonctionner normalement, sinon elle provoquera certaines maladies telles que le cancer et la maladie d'Alzheimer. Par conséquent, la localisation subcellulaire précise des protéines peut être considérée comme l’une des tâches essentielles des sciences de la vie.
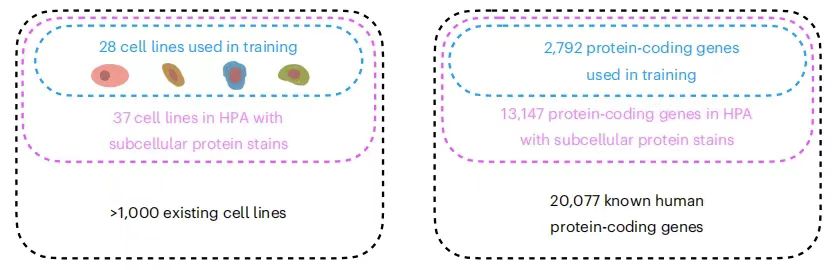
Bien que la localisation spatiale de milliers de protéines dans différentes lignées cellulaires ait été analysée, le nombre de combinaisons protéine-lignée cellulaire qui ont été mesurées jusqu'à présent n'est que la pointe de l'iceberg. Par exemple, le plus grand ensemble de données de localisation subcellulaire actuellement disponible est :L'Atlas des protéines humaines (HPA) fournit la localisation subcellulaire des protéines codées par 13 147 gènes (représentant 651 TP3T de gènes codant pour des protéines humaines connues).Cependant, l’ensemble des données comprenait 37 lignées cellulaires, et chaque protéine a été mesurée dans au plus trois d’entre elles. Dans le même temps, les méthodes expérimentales traditionnelles rendent difficile la détection du nombre total de protéines dans la même cellule en même temps, ce qui entrave sérieusement l’analyse complète des réseaux protéiques complexes et augmente la complexité expérimentale et les risques d’erreur.
De plus, la localisation des protéines n’est pas statique et sa variabilité se produit non seulement entre les lignées cellulaires, mais également entre les cellules individuelles d’une même lignée cellulaire. Les paires de protéines et de lignées cellulaires enregistrées dans les cartes de données existantes ne reflètent que les résultats dans des conditions spécifiques. donc,Même les résultats existants sont difficiles à appliquer directement, et une exploration plus approfondie de la localisation des protéines est nécessaire en fonction des changements environnementaux.
Afin de résoudre la contradiction entre les limites des méthodes de localisation subcellulaire des protéines et la complexité des systèmes biologiques, l’apprentissage automatique devrait être prometteur. Les modèles qui ont été construits et appliqués avec succès aujourd'hui, tels que les modèles basés sur les séquences de protéines et les modèles basés sur les images cellulaires, ont donné de bons résultats dans certains aspects, mais leurs défauts sont également très importants : les premiers ignorent les différences de localisation spécifiques des types de cellules, et les seconds n'ont pas la capacité de généralisation nécessaire pour promouvoir l'étude de protéines inconnues.
Compte tenu de cela,Une équipe de recherche du Massachusetts Institute of Technology et de l'Université Harvard a proposé un cadre de prédiction pour la localisation subcellulaire de protéines inconnues en combinant des séquences de protéines et des images cellulaires, nommé Predictions of Unseen Proteins' Subcellular localization (PUPS). PUPS combine de manière innovante un modèle de langage protéique et un modèle d'inpainting d'image pour prédire la localisation des protéines, lui permettant de fusionner les capacités de généralisation des prédictions de protéines inconnues avec des prédictions spécifiques au type de cellule qui capturent la variabilité cellulaire. Des expériences ont montré que ce cadre peut prédire avec précision la localisation des protéines dans de nouvelles expériences en dehors de l'ensemble de données de formation, possède une excellente capacité de généralisation et une grande précision, et présente un potentiel d'application exceptionnel.
Les résultats de la recherche, intitulés « Prédiction de la localisation subcellulaire des protéines dans des cellules individuelles », ont été publiés dans Nature Methods.
Points saillants de la recherche :
* L'étude proposée combine de manière innovante des modèles de langage protéique et des modèles de rendu d'images, en utilisant des séquences de protéines et des images cellulaires pour prédire la localisation des protéines, compensant ainsi les lacunes des modèles informatiques précédents.
* PUPS est capable de se généraliser à des protéines et des lignées cellulaires inconnues, évaluant ainsi la variabilité de la localisation des protéines entre les lignées cellulaires et entre les cellules individuelles au sein d'une lignée cellulaire, et identifiant les processus biologiques associés aux protéines à localisation variable
* Dans de nouvelles expériences en dehors de l'ensemble de données de formation, PUPS a également démontré sa capacité de prédiction très précise, avec un potentiel d'application exceptionnel et une valeur médicale

Adresse du document :
Ensembles de données : créer des modèles fiables avec les données les plus complètes possibles
L'ensemble de données de formation de PUPS provient du Human Protein Atlas (HPA).L'équipe de recherche a agrégé la 16e version des données HPA dans la 22e version pour collecter autant de données protéiques que possible et garantir l'exhaustivité de l'analyse expérimentale. Comme le montre la figure suivante :
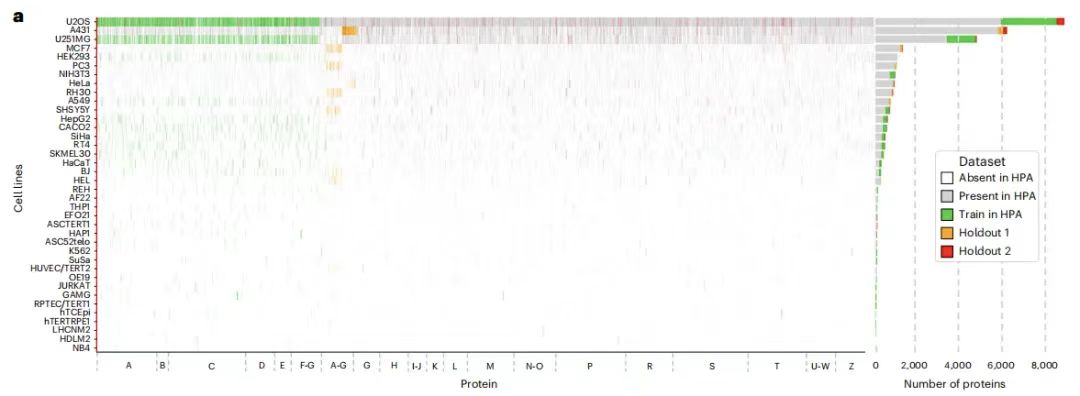
Plus précisément, l'ensemble de données de formation contient 340 553 populations cellulaires avec un total de 8 086 variantes de protéines correspondant à 2 801 gènes dans 37 lignées cellulaires de HPA, dont les noms commencent par les lettres AG. De plus, l'ensemble de données de formation comprend également 10 gènes supplémentaires, dont IHO1, IMPAD1, INKA1, ISPD, ITPRID1, KIAA1211L, KIAA1324, LRATD1, SCYL3 et TSPAN6.
L'ensemble de données de rétention est divisé en deux parties :Une partie est réservée à l'ensemble de données 1,Il contient 36 552 cellules, avec 9 472 variantes de protéines correspondant à 3 312 gènes (dont 2 801 dans l'ensemble d'entraînement), dont les noms commencent également par AG mais proviennent de lignées cellulaires différentes et n'ont aucun chevauchement avec l'ensemble d'entraînement. Entre-temps, l'ensemble de données retenu 1 a été divisé en deux parties et utilisé comme ensemble d'évaluation et ensemble de test, contenant respectivement 11 050 et 25 502 cellules ;L'ensemble de données conservé 2 contient 24 007 cellules, correspondant à 515 gènes.Son nom commence par toutes les lettres de l'alphabet, couvrant de A à Z. Il existe au total 556 variantes de protéines, qui proviennent de nouvelles familles de gènes qui n'apparaissent pas dans l'ensemble d'entraînement et l'ensemble de données réservé1 et peuvent être utilisées pour tester la capacité de généralisation du modèle.
Il convient de noter que les images de la lignée cellulaire BJ ont été conservées à la fois dans l'ensemble d'entraînement et dans l'ensemble de données de maintien 1.
Avant l'expérience, l'équipe de recherche a prétraité les images dans HPA, ce qui comprenait simplement les cinq étapes suivantes :
* Étape 1,Chaque image a été sous-échantillonnée 4 fois et la résolution finale a été réduite à 0,32 μm/pixel afin de réduire la quantité de calcul et de supprimer le bruit haute fréquence ;
* Étape 2,Le flou gaussien (σ=5) et le seuillage d'Otsu ont été combinés pour séparer la zone approximative du noyau cellulaire de l'arrière-plan complexe ;
* Étape 3, utilisez la fonction remove_small_holes pour supprimer les trous d'une zone inférieure à 300 pixels, puis binarisez l'image et supprimez les zones de bruit inférieures à 100 pixels ;
* Étape 4,Le centroïde de chaque noyau cellulaire a été calculé et une zone de 128 × 128 pixels a été découpée avec le centroïde comme ROI d'une seule cellule ;
* Étape 5,Grâce à la normalisation de l'intensité et au filtrage du bruit, une distribution de données standardisée est obtenue et les interférences entre canaux sont réduites.
Architecture du modèle : Combinaison de la séquence protéique et de la représentation d'image pour prédire la localisation subcellulaire des protéines
Le modèle PUPS se compose principalement de deux parties :L'un est utilisé pour apprendre la représentation de la séquence à partir de la séquence d'acides aminés de la protéine ; l'autre est utilisé pour apprendre la représentation de l'image à partir de la coloration iconique de la cellule cible.La représentation de la séquence protéique et la représentation de l'image sont ensuite combinées pour prédire la localisation subcellulaire de la protéine dans les cellules cibles. Le premier permet au modèle d’être généralisé à des prédictions de protéines inconnues, et le second permet au modèle de capturer la variabilité au niveau de la cellule unique et d’obtenir des prédictions de localisation spécifiques au type de cellule. Comme le montre la figure suivante :
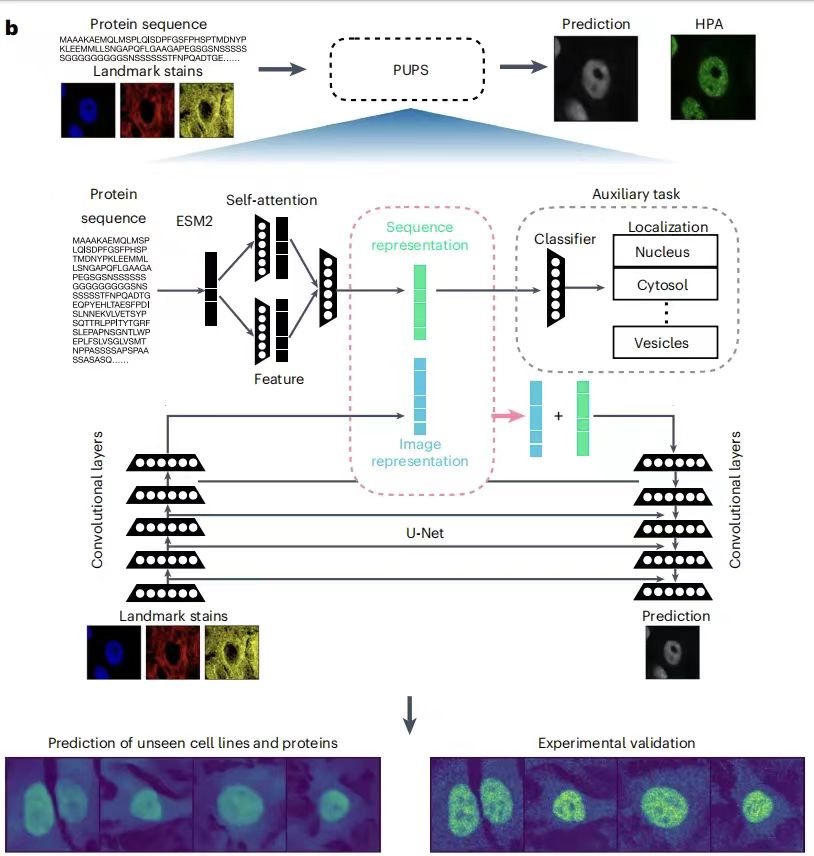
En termes simples,PUPS utilise le modèle de langage protéique ESM-2 (Evolutionary Scale Modeling) pré-entraîné pour extraire les caractéristiques de la séquence protéique et utilise un réseau neuronal convolutif pour apprendre les caractéristiques de l'image de coloration emblématique des cellules. Enfin, les deux parties d’information sont combinées pour prédire la localisation des protéines dans les cellules cibles.Il convient de noter que toutes les parties du modèle sont entraînées simultanément, ce qui contribue à réduire la perte de classification de la pré-tâche et la différence entre les images de protéines prédites et les images de protéines mesurées expérimentalement dans HPA. Tous les paramètres sont optimisés à l'aide de l'optimiseur Adam avec un taux d'apprentissage de 1e-4.
Modèle de langage des protéines
PUPS apprend les représentations de séquences en utilisant un modèle de langage, une couche d'auto-attention et une tâche de pré-formation auxiliaire, puis classe la localisation des protéines en fonction des représentations de séquences apprises.
Plus précisément, l'équipe de recherche a obtenu une représentation initiale d'une variante protéique spécifique en introduisant la séquence N-terminale de 2 000 acides aminés dans le modèle ESM-2 pré-entraîné, générant ainsi un vecteur de 1 280 dimensions pour chaque résidu d'acide aminé, avec un remplissage nul pour les variantes avec moins de 2 000 résidus. Cette limite de longueur de séquence vise à éviter les prédictions biaisées pour les quelques protéines dont la longueur de séquence peut atteindre des dizaines de milliers de résidus. Comme le montre la figure suivante :
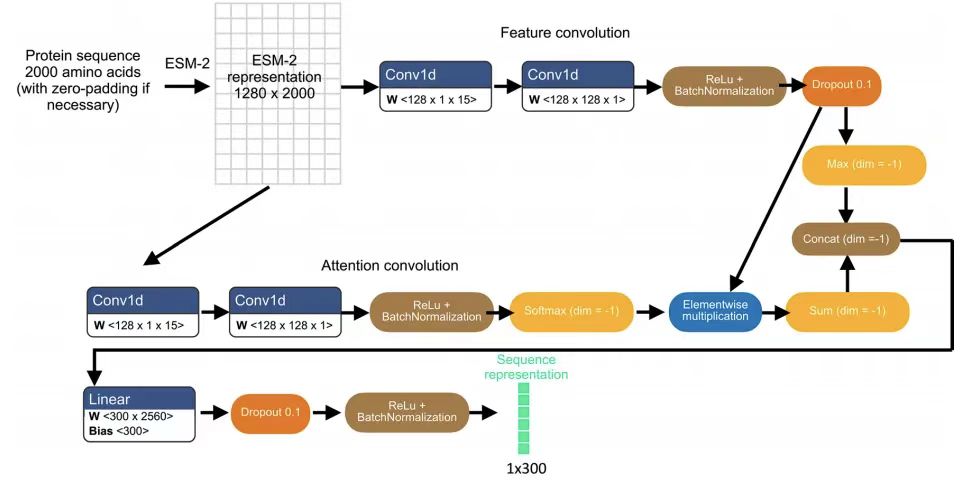
Pour adapter la caractérisation ESM-2 à la prédiction de la localisation des protéines,L’équipe a ensuite adopté une couche d’attention légère de convolutions séparables.Appliquée à la représentation ESM-2, une représentation de séquence à 300 dimensions est finalement obtenue. Cette représentation de séquence protéique est utilisée à la fois pour la pré-tâche auxiliaire de prédiction des étiquettes de localisation et pour la prédiction d'images protéiques en combinaison avec la représentation d'images. La pré-tâche saisit la représentation de la séquence protéique dans une couche de réseau neuronal entièrement connectée pour saisir un vecteur à 29 dimensions représentant la distribution de probabilité parmi 29 étiquettes de localisation de compartiments sous-cellulaires, puis compare la sortie de la pré-tâche avec les compartiments protéiques annotés HPA à l'aide d'une perte d'entropie croisée binaire avec activation sigmoïde.
Modèle de rendu d'image
L'entrée d'image de chaque cellule contient trois canaux d'image de coloration emblématiques : le noyau cellulaire, les microtubules et la coloration du réticulum endoplasmique.Ses dimensions sont de 3 x 128 x 128 et sont centrées au centre du noyau.
Le codage des images est réalisé grâce à 5 couches convolutives séparables.Dimensions finales 16 x 16 x 512 . Chaque couche convolutive est suivie d'une activation leakyRelu, d'une normalisation par lots et de couches de pooling max 2D. La représentation de la séquence protéique est concaténée à toutes les dimensions spatiales de la représentation de l'image cellulaire, puis introduite dans un décodeur d'image U-Net qui apprend différents poids pour chaque canal d'entrée. De plus, le mécanisme de pondération des dimensions spatiales dans le modèle permet de combiner chaque dimension spatiale de la représentation de l'image avec la représentation de la séquence avec des poids différents.
Le décodeur est constitué de 5 couches convolutives séparables.Génère une sortie d'image 1 x 128 x 128, qui est la prédiction d'image protéique pour la cellule correspondante. Des connexions de saut similaires à celles de la segmentation d'image U-Net sont ensuite ajoutées entre la couche de codage qui génère des représentations d'image de coloration de repère et la couche de décodage qui génère des prédictions d'image de protéines à la même profondeur. L’étude a utilisé une fonction de perte d’erreur quadratique moyenne pour entraîner le modèle afin de minimiser la différence entre les images de protéines prédites et les images de protéines mesurées expérimentalement.
Résultats expérimentaux : Obtenir une localisation subcellulaire précise des protéines au niveau de la cellule unique
Afin de vérifier la faisabilité et l’efficacité du modèle, l’équipe de recherche a proposé un certain nombre d’expériences de vérification. PUPS a montré de bonnes performances dans plusieurs tâches, soulignant ses avantages dans la fusion multi-modèles.
Prédire la variabilité de la localisation des protéines entre les lignées cellulaires
Pour évaluer la performance des PUPS dans la quantification de la variabilité de la localisation des protéines parmi les lignées cellulaires,L'équipe de recherche a calculé le rapport nucléaire des protéines pour quantifier la variabilité de localisation et a constaté que les valeurs prédites étaient fortement corrélées avec les données réelles.Le coefficient de corrélation de Pearson pour Holdout 1 est de 0,794 et le coefficient de corrélation de Pearson pour Holdout 2 est de 0,878. Comme le montre la figure suivante :
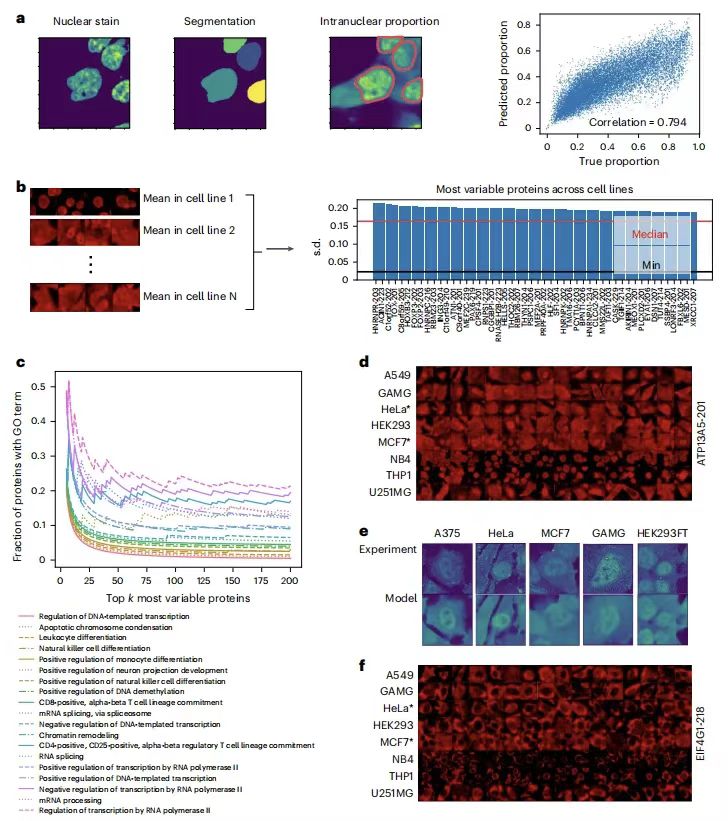
Des analyses ultérieures ont montré que les protéines présentant les plus grands changements de localisation entre les lignées cellulaires étaient liées à des processus biologiques tels que la transcription, la différenciation cellulaire et la régulation de la chromatine. La validation expérimentale de l’ATP13A5 a confirmé l’exactitude des prédictions du modèle. aussi,Le modèle capture les différences de morphologie cellulaire grâce à la coloration de signature et peut déduire la spécificité de la lignée cellulaire de la localisation des protéines sans étiquettes de lignée cellulaire., fournissant une nouvelle méthode pour étudier la régulation spécifique des cellules de la fonction des protéines.
Prédire les différences de localisation des protéines entre les cellules individuelles
Pour évaluer la capacité des PUPS à prédire la variabilité de la localisation des protéines entre les cellules individuelles d’une même lignée cellulaire, l’équipe de recherche a calculé la variance du rapport nucléaire des protéines dans toutes les cellules individuelles de chaque lignée cellulaire.Les résultats ont montré que le classement de prédiction de la variabilité unicellulaire pour chaque paire protéine-lignée cellulaire était très cohérent avec les données réelles.Par exemple, le taux de chevauchement des 500 premières paires de variantes élevées dans Holdout 2 dépassait 60%, et la distribution du rapport intranucléaire prédite était cohérente avec les résultats réels, éliminant ainsi l'influence des erreurs de prédiction.
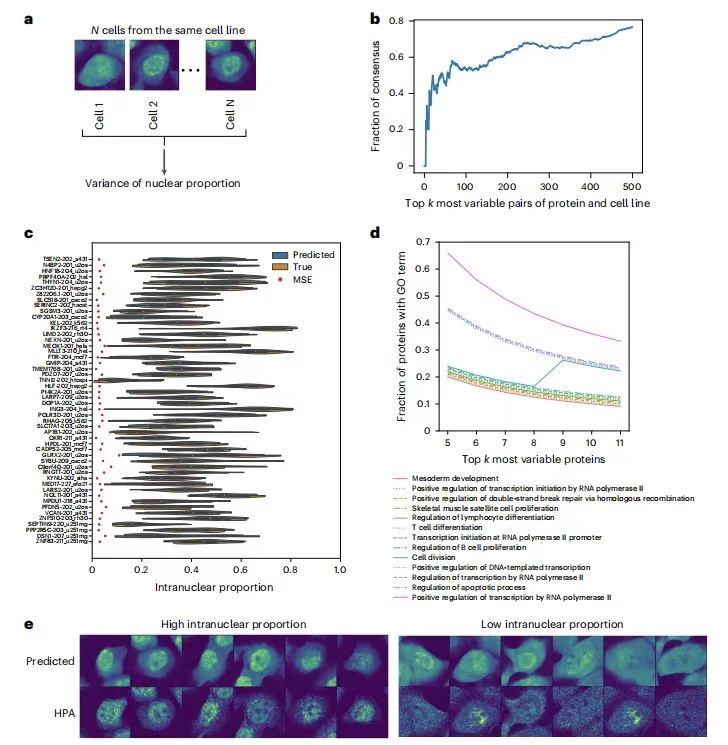
De plus, l’analyse de l’ontologie génétique (GO) a montré que des protéines très variables étaient liées à des processus tels que la division cellulaire, la transcription, la réparation des cassures double brin et l’apoptose. aussi,Le modèle capture les caractéristiques morphologiques grâce à des images de marquage cellulaire, indiquant que la variabilité unicellulaire n'est pas seulement aléatoire mais également liée aux caractéristiques morphologiques cellulaires.Fournit une nouvelle perspective pour expliquer le mécanisme de l’hétérogénéité unicellulaire.
Validation des PUPS dans de nouvelles expériences en dehors des données d'entraînement
Afin de vérifier la capacité d’ubiquitination des PUPS à prédire la localisation des protéines dans de nouveaux environnements expérimentaux, l’équipe de recherche a sélectionné 9 protéines à vérifier dans 5 lignées cellulaires. Comme le montre la figure suivante :
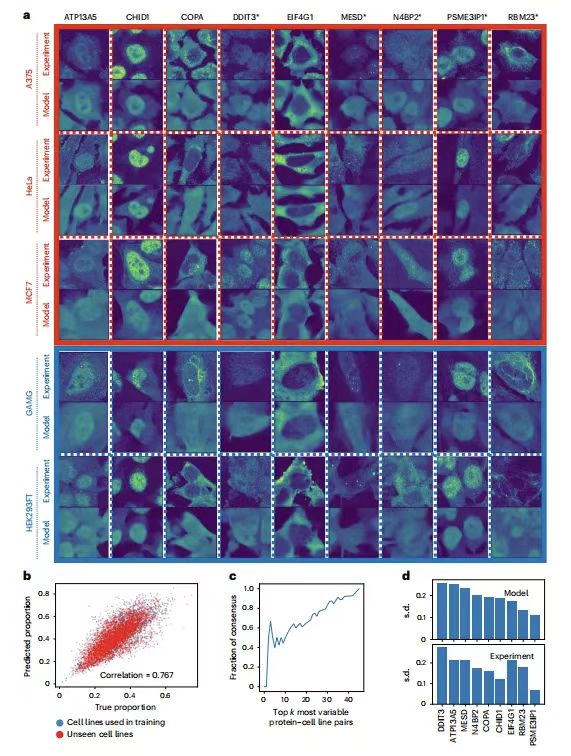
ATP13A5, CHID1, COPA, MESD et RBM23 sont les protéines présentant la plus grande variation parmi les lignées cellulaires, et elles ont toutes des termes GO différents ; DDIT3 et N4BP2 sont les protéines présentant la plus grande variation dans les cellules individuelles d’une lignée cellulaire ; EIF4G1 et PSME3IP1 sont les protéines présentant le moins de variations parmi les lignées cellulaires, la première étant censée être principalement située à l'extérieur du noyau et la seconde étant censée être principalement située à l'intérieur du noyau. Parmi les cinq lignées cellulaires, à l'exception de A375, les autres HeLa, MCF7, GAMG et HEK293FT sont incluses dans le HPA.
Les résultats montrent queLes images protéiques prédites par PUPS sont visuellement similaires à celles mesurées expérimentalement.Le rapport protéique nucléaire de chaque cellule individuelle calculé à l'aide de l'image protéique prédite est étroitement corrélé au rapport calculé à partir de l'image mesurée expérimentalement, avec un coefficient de corrélation de Pearson de 0,767. Cela montre queLes PUPS peuvent être utilisés pour prédire quantitativement la localisation de protéines qui n’ont pas été mesurées expérimentalement auparavant ou utilisées dans des atlas de formation.
PUPS apprend des représentations significatives des protéines et des cellules
Des expériences démontrent que la capacité du PUPS à prédire la localisation des protéines dans des protéines et des lignées cellulaires inconnues provient de l’apprentissage de représentations significatives de séquences de protéines et d’images de repères cellulaires.
L'équipe de recherche a cartographié les représentations de séquences protéiques de 40 622 formes de protéines correspondant à 12 614 gènes, et les protéines avec des localisations similaires avaient tendance à avoir des représentations de séquences similaires. Pour démontrer davantage que le modèle peut identifier des modèles de séquences protéiques significatifs et prédire la localisation, l’équipe de recherche a utilisé la méthode Positional Shapley pour calculer l’importance de chaque résidu d’acide aminé dans une protéine spécifique pour prédire les étiquettes de chaque compartiment cellulaire. Par exemple, il a expliqué avec succès la variabilité prédite de la localisation nucléaire de N4BP2, ce qui est cohérent avec les rapports selon lesquels le domaine CUE peut modifier la localisation sous-cellulaire par la liaison à l'ubiquitine.
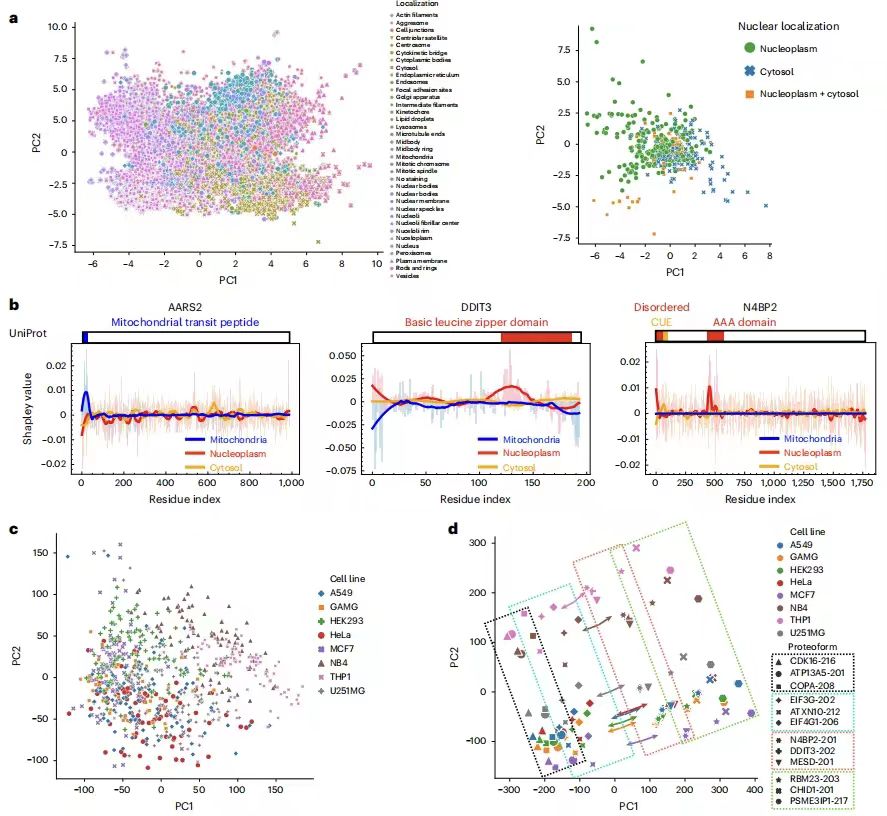
En plus d'identifier des motifs de séquences protéiques significatifs,L’équipe de recherche a également démontré que PUPS apprend des représentations significatives de cellules individuelles à partir de la coloration de la signature cellulaire.Il visualise les représentations d'images de cellules individuelles apprises à partir de la coloration des repères et constate que les cellules individuelles de la même lignée cellulaire ont des représentations d'images similaires même si l'étiquette de la lignée cellulaire n'est pas entrée dans le modèle. La représentation conjointe des protéines et des images de repères cellulaires préserve la séparation entre les lignées cellulaires et les protéines, tandis que l'ordre des différentes protéines au sein de chaque lignée cellulaire est similaire d'une lignée cellulaire à l'autre. Étant donné le centroïde de chaque lignée cellulaire dans l'espace de représentation conjoint, les vecteurs du centroïde vers une protéine spécifique sont pour la plupart parallèles sur toutes les lignées cellulaires, c'est-à-dire que, étant donné la représentation de la séquence, la prédiction d'une image pour une protéine spécifique nécessite de se déplacer dans la même direction dans l'espace de représentation quelle que soit la lignée cellulaire.Cela explique la capacité des PUPS à généraliser à des protéines et des lignées cellulaires inconnues en apprenant des représentations significatives d'images de protéines et de cellules.
aussi,Les PUPS peuvent également prédire les effets des mutations causant des maladies sur la localisation des protéines.Par exemple, des études de mutation sur les protéines mitochondriales codées dans le noyau SDHD et ETHE1 ont montré que les mutations SDHD entraînent une augmentation de leur rapport de localisation nucléaire, ce qui est cohérent avec le mécanisme d’instabilité du génome nucléaire dans la maladie ; Les mutations ETHE1 montrent une augmentation du rapport de localisation cytoplasmique, qui est associée à des anomalies connues de la navette nucléaire-cytoplasmique. Ces résultats suggèrent que les PUPS peuvent fournir de nouveaux indices pour étudier les mécanismes de la maladie en analysant les effets de la variation de séquence sur la localisation.
Nouvelle solution pour la prédiction de la localisation subcellulaire des protéines
Comme mentionné ci-dessus, la prédiction de la localisation subcellulaire des protéines est d’une grande importance en bioinformatique et en recherche biologique. PUPS offre un moyen d'intégrer des informations multimodales, ce qui ajoute une touche forte à la recherche dans ce domaine. Parallèlement, après des décennies de développement, la recherche dans ce domaine a donné lieu à une grande variété de résultats.
Une équipe de l'University College Dublin en Irlande a publié une étude dans le Computational and Structural Biotechology Journal.Il présente une variété de méthodes informatiques pour la prédiction de la localisation subcellulaire des protéines, notamment des méthodes basées sur les séquences, les annotations, les hybrides et les méta-prédictions. L'article classe et présente également des outils de prédiction de localisation subcellulaire par eucaryotes, procaryotes, virus et plusieurs catégories.Les outils de prédiction eucaryotes incluent mLASSO-Hum, DeepPSL, etc., et les outils de prédiction procaryotes incluent PRED-LIPO, etc. En concevant une carte de classification de l'apprentissage automatique et de l'apprentissage profond couvrant 7 domaines principaux et 28 sous-catégories, cette étude fournit une taxonomie d'outils de prédiction mono-catégorie et multi-catégories, ce qui permet aux utilisateurs de trouver plus facilement des méthodes et des outils de prédiction. L’article a été publié sous le titre « Outils de prédiction de la localisation subcellulaire des protéines ».
* Adresse du papier :
https://www.sciencedirect.com/science/article/pii/S2001037024001156
Le 12 avril, le groupe de recherche de Yang Li à l'Institut des sciences biomédicales de l'Université Fudan et le groupe de recherche de Dong Nanqing au Laboratoire d'intelligence artificielle de Shanghai ont collaboré pour publier un article de recherche en ligne intitulé « Modèle génératif profond pour la localisation subcellulaire des protéines » dans la revue Briefings in Bioinformatics.L'étude a également développé un modèle d'apprentissage profond génératif deepGPS avec des capacités de traitement multimodal basées sur le modèle de langage protéique ESM2 et le framework U-Net.
Selon les rapports, deepGPS peut recevoir des séquences de protéines et des images de noyaux cellulaires en entrée et générer des étiquettes de texte et des images de distribution de localisation de protéines. Il s'agit d'un nouveau modèle multimodal « texte-image » qui prend en charge la prédiction de la localisation subcellulaire des protéines.
* Adresse du papier :
https://doi.org/10.1093/bib/bbaf152
À mesure que l'intégration de l'intelligence artificielle et de la recherche biologique s'accélère, des expériences innovantes connexes émergent constamment, brisant progressivement les inconvénients des méthodes traditionnelles, obtenant « le meilleur des deux mondes » ou même des performances « parfaites », favorisant ainsi le développement rapide de la bioinformatique.








