Command Palette
Search for a command to run...
Pour La Première Fois, Une Équipe De l'Université De Columbia a Proposé PXRDnet Pour Analyser Les Nanocristaux De Bout En Bout Et a Analysé Avec Succès 200 Nanocristaux Simulés complexes.

La découverte et l’application de la diffraction des rayons X (DRX) constituent une étape importante dans le développement de la cristallographie, car cette technologie permet aux gens d’acquérir une compréhension approfondie de la microstructure des cristaux, ce qui à son tour stimule le progrès de la science des matériaux et de la civilisation humaine dans son ensemble. Cependant, lorsque les méthodes traditionnelles rencontrent des nanocristaux en poudre composés de minuscules particules, les résultats souhaités ne se produisent pas.
En raison de la taille limitée des nanocristaux (généralement inférieure à 1000 Å),Le pic de Bragg dans son diagramme de diffraction des rayons X montre un élargissement évident.Cela entraîne une dégradation substantielle et sérieuse des informations structurelles, ce qui pose un énorme défi pour résoudre avec précision sa structure cristalline. De plus, la difficulté d’obtenir des échantillons monocristallins purs dans des situations réelles augmente encore la difficulté de l’analyse structurelle. L’analyse de la structure des nanocristaux est également devenue un « problème vieux d’un siècle » qui tourmente la communauté scientifique des matériaux depuis cent ans.
Pour répondre à ce problème, des chercheurs de l’Université de Columbia et de l’Université de Stanford ont proposé une méthode d’analyse de structure d’intelligence artificielle générative PXRDnet basée sur un modèle de diffusion.Le modèle utilise 45 229 structures cristallines connues comme données d’apprentissage et introduit des connaissances statistiques préalables.Même avec seulement la formule chimique et le modèle de diffraction de poudre élargi de taille limitée et peu informatif comme conditions, PXRDnet a pu résoudre avec succès 200 nanocristaux simulés de symétrie et de complexité variables.Les structures des sept systèmes cristallins sont incluses, jusqu'à une taille de 10 Å.Les résultats expérimentaux montrent que le modèle peut identifier avec succès et de manière vérifiable 4 candidats structurels sur 5, avec une erreur moyenne de seulement 7% après mesure par le facteur r de raffinement de Rietveld.
La recherche connexe a été publiée dans Nature Materials sous le titre « Solutions de structure ab initio à partir de données de diffraction de poudre nanocristalline via des modèles de diffusion ».
Points saillants de la recherche :
* Cette réalisation a résolu le problème de longue date de l'analyse de la structure des nanocristaux dans la communauté des sciences des matériaux et a fourni un outil d'analyse d'intelligence artificielle efficace, qui devrait promouvoir des applications innovantes dans les nanotechnologies, la biomédecine, le stockage d'énergie, les appareils électroniques et d'autres domaines.
* Cette méthode dépasse considérablement les limites d'applicabilité des méthodes traditionnelles et obtient des solutions candidates proches de la structure réelle dans de nombreux cas
* L'étude a proposé l'ensemble de données de référence MP-20-PXRD (y compris les matériaux stables avec moins de 20 atomes dans le projet Matériaux et leurs données de diffraction simulées), et a rendu le code et l'ensemble de données publics, fournissant une norme unifiée pour les recherches ultérieures

Adresse du document :
https://go.hyper.ai/r1K6b
Base de données de matériaux en ligne du projet Matériaux :
https://go.hyper.ai/2gCe9
Ensemble de données : Ensemble de données de référence MP-20-PXRD proposé
Pour obtenir un modèle efficace, les chercheurs ont fourni un ensemble de données de référence appelé MP-20-PXRD pour la formation de bout en bout de PXRDnet.
Plus précisément, les chercheurs ont utilisé l’ensemble de données MP-20 du projet Matériaux.L'ensemble de données se compose de matériaux échantillonnés à partir de la base de données Materials Project avec un maximum de 20 atomes dans la cellule unitaire.Les chercheurs ont ensuite utilisé le package pymatgen pour simuler les modèles de diffraction de poudre de toutes les structures du MP-20.
Base de données de matériaux en ligne du projet Matériaux :
https://go.hyper.ai/2gCe9
Les simulations ont utilisé un rayonnement Cu Kα avec une plage Q de 0 à 8,1568 Å⁻¹.
L'ensemble de données MP-20-PXRD contient 45 229 matériaux.Les ratios de 90%, 7,5% et 2,5% sont utilisés pour la formation, la vérification et les tests. Il convient de mentionner que l'ensemble de données MP-20-PXRD a été ouvert en open source et que les chercheurs espèrent l'utiliser pour inspirer les « retardataires » à explorer davantage de nouvelles solutions pour l'analyse de la structure des nanocristaux.
Architecture du modèle : basée sur CDVAE, introduction du régresseur PXRD
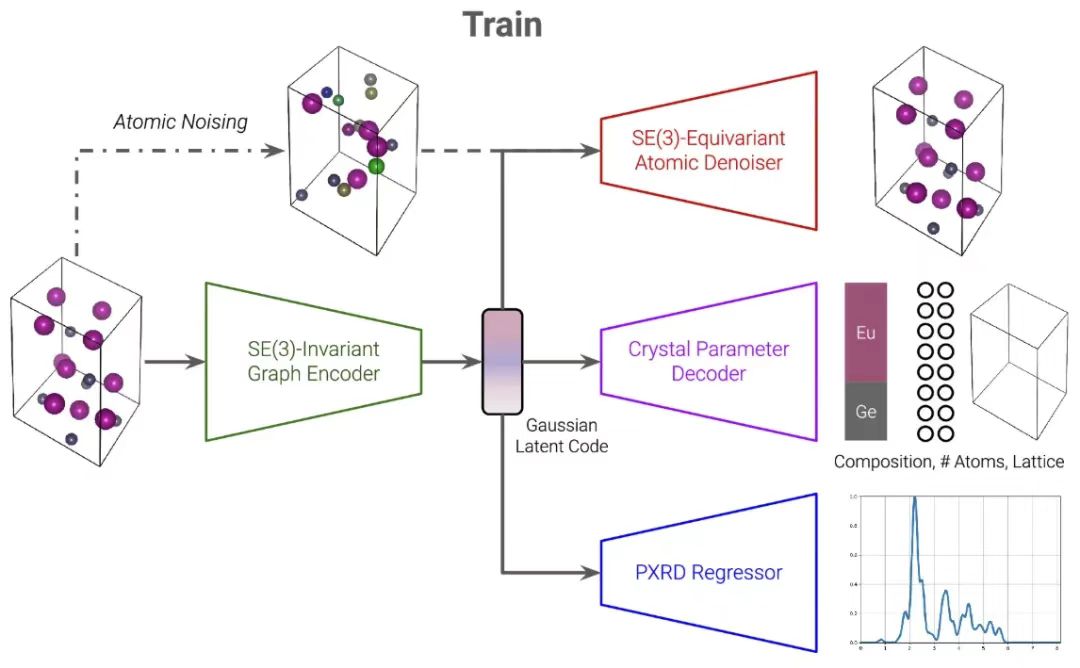
Le modèle PXRDnet est conçu sur la base de l'architecture CDVAE.Il se compose principalement de trois branches principales, à savoir la branche de débruitage atomique, la branche d'autoencodeur variationnel (VAE) et le régresseur PXRD.Ils sont connectés via un code latent gaussien partagé. Cette approche permet à PXRDnet de générer avec précision des candidats de structure de matériaux qualifiés à partir d'un modèle PXRD et d'une formule chimique, offrant de nouvelles perspectives sur l'analyse de la structure des nanomatériaux.
Développement du squelette basé sur CDVAE
Lors de l'introduction de PXRDnet, nous devons mentionner le modèle CDVAE, qui est la base de la création du premier.CDVAE est un modèle de génération de structure matérielle.Il s'inspire des autoencodeurs variationnels et des réseaux de diffusion de débruitage et constitue un modèle génératif qui apprend à décompresser les données du bruit.
Pour comprendre la décomposition des composants VAE et de diffusion, les chercheurs ont réalisé que la cellule unitaire d'un matériau peut être représentée par quatre composants : la composition chimique, le nombre d'atomes, les paramètres du réseau et les coordonnées atomiques.
La première branche de CDVAE utilise VAE pour traiter les trois premiers composants.L'encodeur est DimeNet, un réseau neuronal graphique invariant SE(3) qui mappe la représentation graphique du matériau à une représentation latente z. La représentation graphique est modifiée en un multigraphe dirigé pour refléter la périodicité inhérente du matériau. Les chercheurs ont ensuite régularisé la représentation latente z dans une distribution gaussienne multivariée en utilisant la perte de divergence de Kullback-Leibler, puis ont décodé la composition chimique, le numéro atomique et les paramètres de réseau de z.
Chaque prédiction est générée par un perceptron multicouche paramétré par cristal (MLP) distinct qui reçoit le code latent z.z sera utilisé comme représentation matérielle dans toutes les autres branches du modèle suivant.
La deuxième branche de CDVAE utilise la diffusion de débruitage pour traiter les composants via un réseau de score conditionné par le bruit.On suppose que le nombre d’atomes composants et les paramètres du réseau sont fixes. Le processus direct utilise un bruit gaussien multivarié pour perturber les coordonnées atomiques et les espèces atomiques. Le processus inverse est paramétré à l'aide de GemNet, un réseau neuronal graphique SE(3)-équivariant. Ce processus est conditionné par le code latent z décrit ci-dessus, qui est la base de son fonctionnement normal.
Il convient de mentionner queLe processus inverse consiste essentiellement à prédire comment débruiter les coordonnées atomiques et les espèces perturbées via la dynamique de Langevin.Faites-les se déplacer vers leurs véritables emplacements et restaurez-les dans leur véritable espèce. De même, la représentation du graphe de sortie est un multigraphe orienté, compatible avec la périodicité du matériau.
Dans la phase de génération, CDVAE échantillonne d'abord un code latent z ≈ N (0, I) à partir d'une distribution gaussienne multivariée.Le perceptron multicouche à paramètres cristallins est utilisé pour le décoder et obtenir la composition chimique du composant, le numéro atomique et les paramètres du réseau, qui peuvent être utilisés pour initialiser une cellule unitaire, où les positions atomiques sont également sélectionnées aléatoirement parmi N (0, I). Les positions et types atomiques sont ensuite optimisés grâce au processus de débruitage d'image équivariante Langevin Dynamics SE (3). Pendant tout le processus de débruitage, les paramètres du réseau et le numéro atomique restent inchangés et le matériau résultant est finalement obtenu.
Régresseur PXRD spécialement conçu
De plus, dans cette étude, le modèle de diffraction des rayons X sur poudre (PXRD) a été défini comme la propriété souhaitée à prédire, les chercheurs ont donc conçu un régresseur PXRD Fψ, qui transforme la représentation matérielle potentielle z∈R256 Mappé sur un vecteur y∈R512, c'est-à-dire la caractérisation estimée de l'espace Q du motif PXRD du matériau.
Le régresseur PXRD est paramétré par une architecture inspirée de DenseNet.Cette architecture étend le réseau neuronal convolutif traditionnel.Le régresseur est basé sur la conception de CrystalNet, avec une architecture densément connectée avec une entrée et une sortie unidimensionnelles. Plus précisément, pour une profondeur donnée dans le réseau,DenseNet regroupe les représentations de données intermédiaires précédentes en tant qu'entrée de la couche convolutive suivante.Comme le montre la figure ci-dessous.
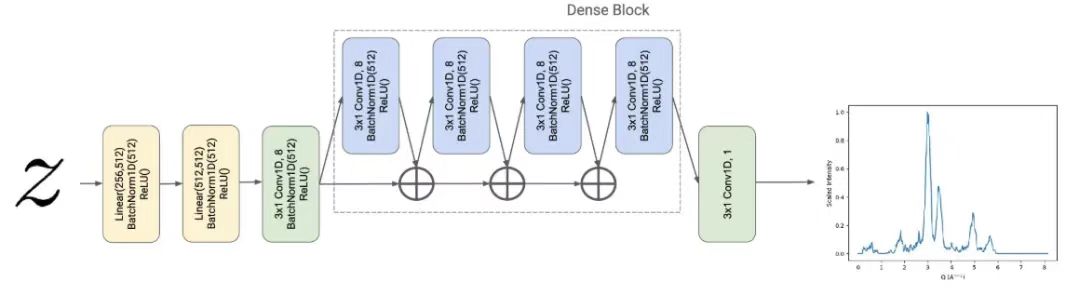
Des recherches ont montré que DenseNet réduit le problème de gradient de disparition et obtient d’excellents résultats sur les tests de vision par ordinateur standard.
Résultats expérimentaux : potentiel d'application dans le monde réel
En règle générale, les nanostructures sont définies comme des cristaux d'une taille inférieure à 1 000 Å, mais pour tester l'efficacité de la méthode proposée, les chercheurs ont réduit la taille des cristaux de deux ordres de grandeur, en simulant la méthode PXRD avec des tailles de cristaux de 10 Å et 100 Å en utilisant une méthode de filtrage mathématique basée sur l'analyse de Fourier. Comme prévu,Le cas 10 Å montre un élargissement de pic plus important que le cas 100 Å, indiquant que la dégradation de l'information est plus confirmée.Comme le montre la figure ci-dessous.
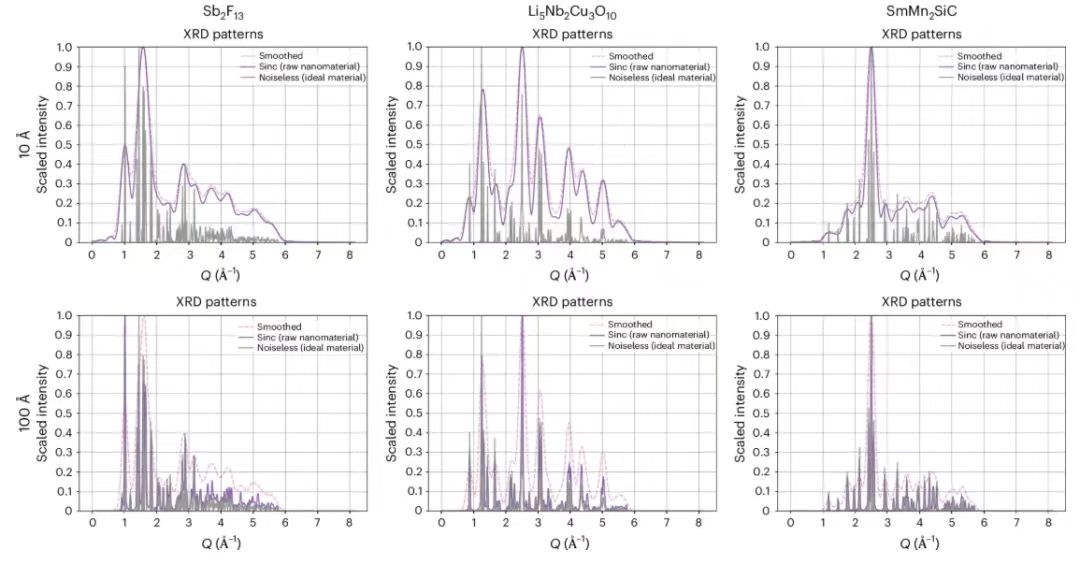
Cette image montre comment les chercheurs ont simulé l’effet du rétrécissement à l’échelle nanométrique sur les pics PXRD à l’aide du filtrage sinc². Parmi eux, la ligne grise représente le mode idéal et la ligne violette représente le pic PXRD qui s'élargit après le traitement.Pour améliorer les performances du modèle, les chercheurs ont appliqué un filtre gaussien supplémentaire après le filtre sinc.Bien que cela augmente l’élargissement des pics de diffraction, cela peut éliminer efficacement les ondulations prononcées causées par le filtrage. L'axe horizontal représente la taille du vecteur de diffusion en Å⁻¹, et l'axe vertical représente l'intensité de diffraction mise à l'échelle, où 1 représente la valeur d'intensité maximale.
Ensuite, les chercheurs ont présenté la prédiction de la structure PXRDnet, comme indiqué ci-dessous. La colonne la plus à gauche montre la structure cristalline réelle, et les autres colonnes montrent les structures cristallines reconstruites de nanocristaux avec des diamètres de 10 Å et 100 Å simulés par PXRDnet dans le modèle PXRD après raffinement de Rietveld.
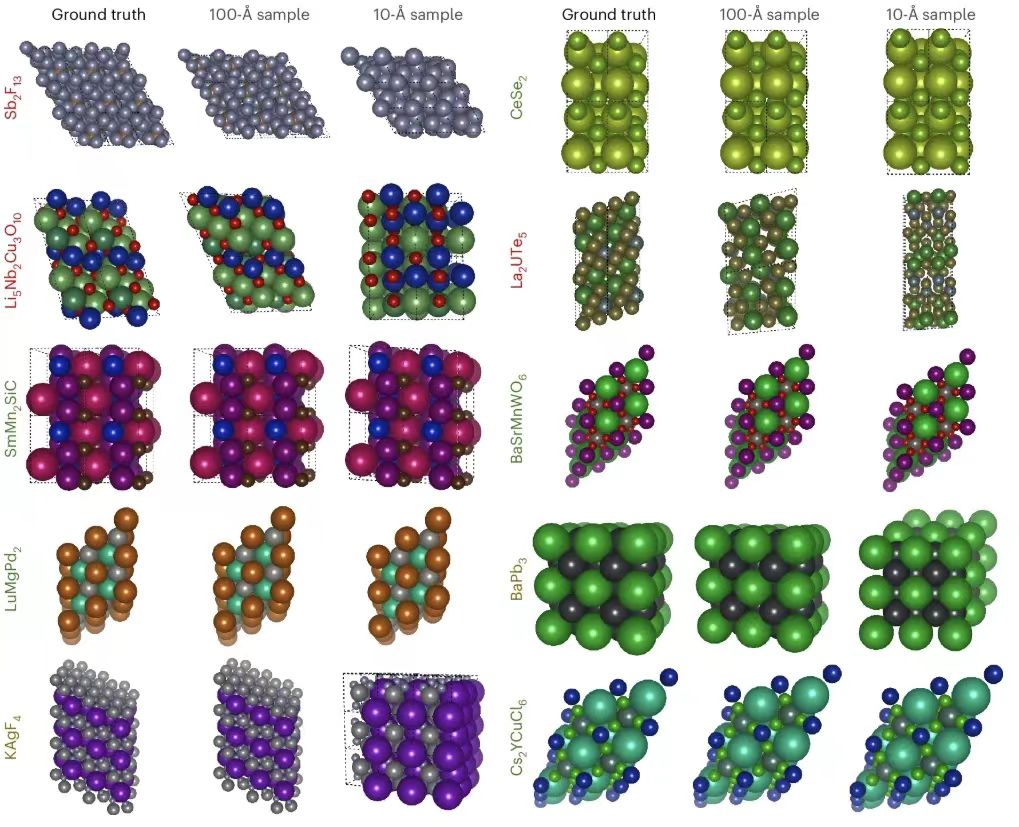
Les résultats montrent que PXRDnet fonctionne bien dans l’analyse de la structure des matériaux de diverses compositions chimiques inorganiques.Les performances sont légèrement meilleures à la taille de cristal de simulation de 100 Å, mais restent excellentes à la taille de cristal de simulation plus difficile de 10 Å.Par exemple, PXRDnet peut capturer avec succès la forme cristalline de matériaux tels que Cs₂YCuCI₆ et SmMn₂SiC, et il peut également capturer avec succès la symétrie de matériaux tels que Cs₂YCuCI₆ et BaSrMnWO₆. De plus, même dans certains cas extrêmes, comme la défaillance de Li₅Nb₂Cu₃O₁₀ ou de Sb₂F₁₃, PXRDnet peut toujours fournir une référence précieuse pour les expériences.
La figure ci-dessous montre la comparaison du modèle PXRD réel, du modèle prédit d'origine par PXRDnet et du modèle après raffinement de Rietveld, qui montre le degré de concordance entre le modèle prédit et les données réelles, et vérifie la nécessité de Rietveld, qui peut améliorer efficacement la précision de prédiction du modèle. Par exemple, à 100 Å, la différence prédite pour Sb₂F₁₃ était de 0,681, qui a été réduite à 0,019 après affinement (AI+Rietveld).
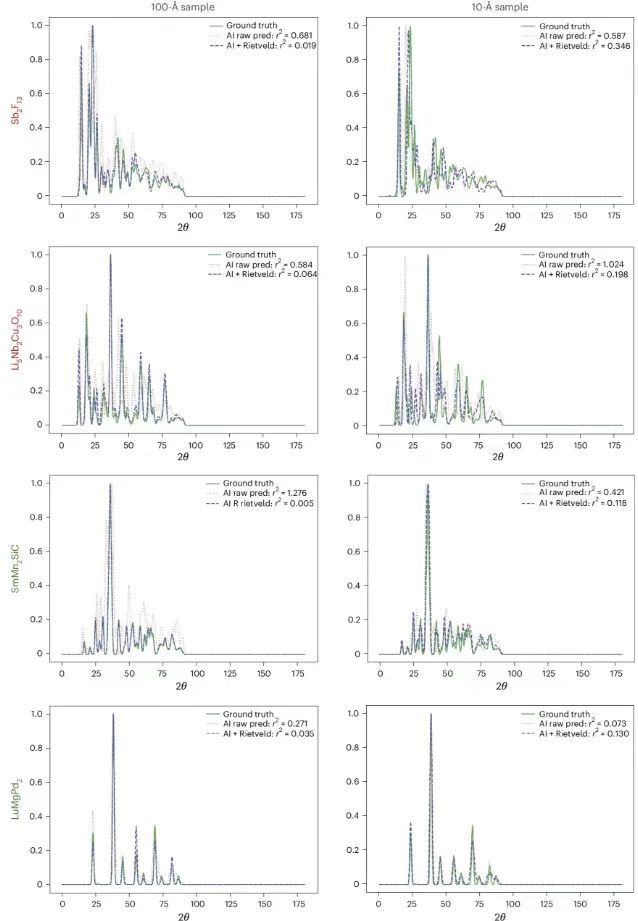
Comparaison du véritable modèle PXRD, du modèle prédit par PXRDnet d'origine et du modèle après raffinement de Rietveld
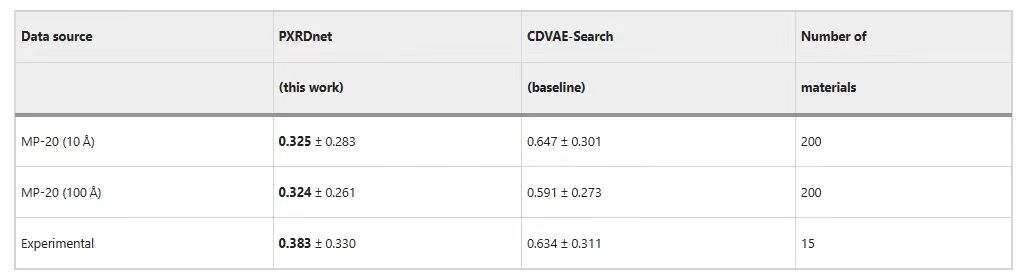
Le tableau suivant montre que PXRDnet peut reconstruire avec succès les matériaux du MP-20.Par rapport à la base de référence CDVAE-Search, les résultats de prédiction de PXRDnet sont plus remarquables.
Pour améliorer encore les résultats, les chercheurs ont effectué un raffinement de Rietveld sur 20 structures sélectionnées uniformément et résolues par PXRDnet, en sélectionnant les 10 meilleures entrées candidates pour chaque structure. Comme le montre la figure ci-dessous.
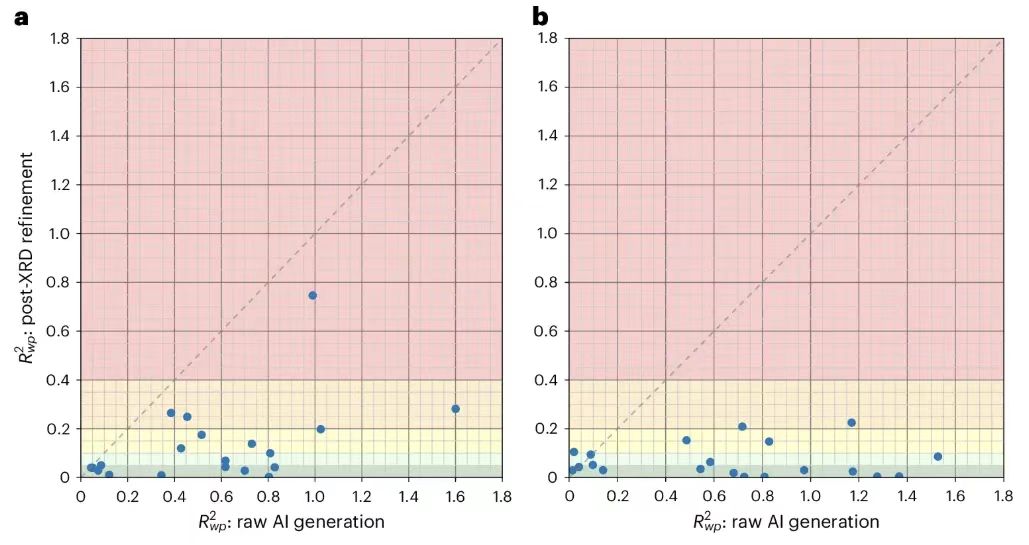
Les résultats montrent queLe raffinement de Rietveld s'est avéré particulièrement efficace pour les tests à 100 Å, qui présentent des pics de Bragg plus nets, avec 18 des 20 structures testées tombant en dessous de 20% et 15 tombant en dessous de 10%.Cela montre que malgré quelques problèmes mineurs, PXRDnet est toujours capable de produire systématiquement des résultats proches de la structure réelle, et la structure correcte peut être obtenue avec une intervention humaine appropriée dans chaque cas.
Enfin, les chercheurs ont vérifié expérimentalement les performances du test PXRD, avec des données de la base de données IUCr. Comme le montre la figure ci-dessous.
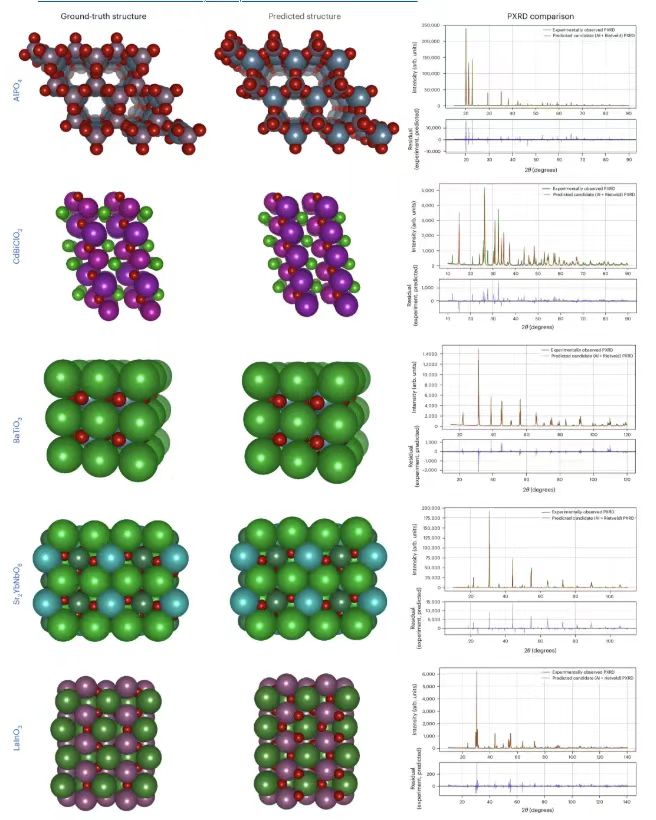
La colonne la plus à gauche montre les structures de référence, basées sur les modèles PXRD observés expérimentalement obtenus à partir de la base de données IUCr, le milieu montre les structures prédites par PXRDnet, et le côté droit montre la comparaison entre le PXRD simulé TOPAS (v.7) et le PXRD réellement observé expérimentalement.Les résultats montrent que PXRDnet surmonte l’écart entre la simulation et la réalité et ses résultats sont comparables à ceux obtenus à partir de données simulées en termes d’analyse visuelle et de mesures quantitatives, démontrant le potentiel du modèle proposé pour une application dans des scénarios du monde réel.
L'IA et la science des matériaux s'associent pour résoudre des problèmes vieux de plusieurs siècles
L’introduction de PXRDnet a résolu un problème vieux d’un siècle dans la communauté des sciences des matériaux. Comme l’indique l’article, la méthode, comme toute solution structurelle, n’est pas efficace à 100 %, mais elle fournit une méthode candidate pour explorer l’élucidation structurelle, ouvrant ainsi davantage de portes vers le succès.
Bien sûr, le succès de PXRDnet ne s’est pas produit du jour au lendemain, mais est le résultat d’une exploration continue en s’appuyant sur les épaules de géants. À l’intersection de l’intelligence artificielle et des nanomatériaux, d’innombrables chercheurs scientifiques travaillent constamment sur des avancées décisives.
Par exemple, la recherche publiée par le MIT, l'Université de Stanford et d'autres équipes sur « Détermination de la structure cristalline à partir de motifs de diffraction de poudre avec apprentissage automatique génératif »Nous présentons ici un modèle d’apprentissage automatique génératif révolutionnaire capable de résoudre des structures cristallines à partir de données PXRD expérimentales réelles.Dans les expériences, les chercheurs ont prédit les structures de 134 modèles expérimentaux à partir de la base de données RRUFF et de milliers de modèles simulés à partir du projet Matériaux, avec des taux de correspondance de modèles atteignant respectivement les taux de pointe 42% et 67%.
Adresse du document :
https://pubs.acs.org/doi/10.1021/jacs.4c10244
En outre, des équipes de l’Académie chinoise des sciences, de l’Université Jiaotong de Shanghai, de l’Université Tsinghua et de l’Université Renmin de Chine ont également publié des recherches connexes.Nous proposons un réseau neuronal de bout en bout, PXRDGen, qui peut déterminer la structure cristalline en apprenant la distribution structurelle des cristaux expérimentalement stables et leurs modèles PXRD.La structure de précision atomique a été extraite des données PXRD. Le modèle hérite d'un encodeur XRD pré-entraîné, d'un générateur de structure basé sur la diffusion/le flux et d'un module de raffinement Rietveld, et peut atteindre avec précision une résolution de structure en quelques secondes seulement. La recherche connexe a été publiée sous le titre « Détermination de la structure cristalline par diffraction des poudres à l’aide de modèles génératifs ».
Adresse du document :
https://arxiv.org/abs/2409.04727
En résumé, l’exploration de PXRDnet et d’autres méthodes a permis à la communauté des sciences des matériaux de passer des méthodes traditionnelles à l’intégration croisée de l’intelligence artificielle et de la science des matériaux. Il a non seulement réalisé des avancées substantielles et résolu les problèmes rencontrés par la communauté scientifique des matériaux, mais a également fourni de nouvelles idées et méthodes pour les recherches ultérieures, insufflant une nouvelle vitalité au développement futur de la science des matériaux.








