Command Palette
Search for a command to run...
Sélectionnée Pour l'ICLR 2025 Oral, l'équipe De Zhou Hao De Tsinghua AIR a Proposé Un Nouveau Paradigme De Pré-entraînement Des Protéines Pour Déchiffrer l'évolution Des Familles De Protéines

Le groupe de recherche AIR GenSI de l'Université Tsinghua et l'École de pharmacie de l'Université Tsinghua ont proposé conjointement un outil de modélisation générative spécifique à la famille de protéines - ProfileBFN (Profile Bayesian Flow Network). ProfileBFN étend le réseau de flux bayésien discret du point de vue des profils d'alignement de séquences multiples (MSA) pour obtenir une conception efficace de la famille de protéines. Les résultats empiriques montrent queTout en générant des protéines familiales diverses et nouvelles, ProfileBFN est capable de capturer avec précision les caractéristiques structurelles de la famille.

Les résultats associés ont été intitulés « Steering Protein Family Design through Profile Bayesian Flow » et ont été sélectionnés comme article oral dans l'ICLR 2025. Dans le même temps, une autre réalisation de l'équipe, CrysBFN, a également été sélectionnée pour ICLR 2025 Spotlight. Le titre du document de recherche est « Un flux bayésien périodique pour la génération de matériaux ».
Lors de la dernière session, l'équipe a proposé le réseau de flux bayésien géométrique GeoBFN, et les résultats associés ont été sélectionnés pour l'oral ICLR 2024 sous le titre « Modélisation générative unifiée de molécules 3D avec des réseaux de flux bayésiens ».

Lien vers l'article :
Le projet open source « awesome-ai4s » rassemble plus de 200 interprétations d'articles AI4S et fournit des ensembles de données et des outils massifs :
https://github.com/hyperai/awesome-ai4s
Alignement de séquences multiples : la pierre angulaire de la prédiction de la structure des protéines
L'alignement de séquences multiples (MSA) fait référence au processus d'alignement de trois séquences biologiques ou plus (ADN, ARN ou protéines). L'alignement de séquences multiples permet de découvrir et d'identifier des régions similaires en raison de relations fonctionnelles, structurelles ou évolutives, offrant une perspective plus complète sur les relations entre les macromolécules biologiques.
Ces dernières années, l’utilisation des informations MSA est devenue un élément important de la conception des protéines. Dans les travaux marquants tels qu'AlphaFold et ESM, il existe des modules spéciaux codant les informations MSA :
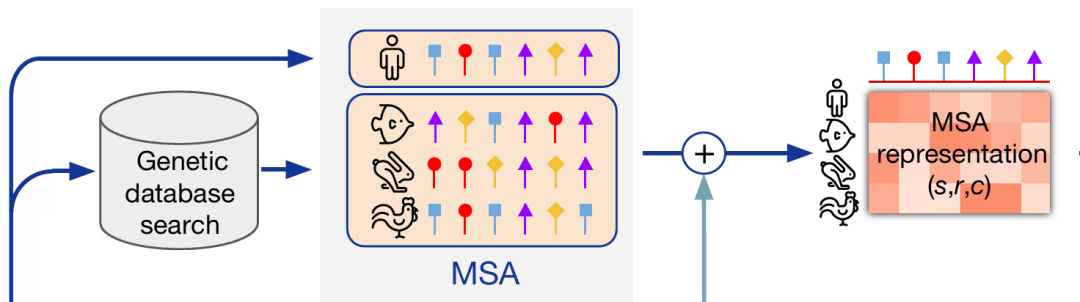
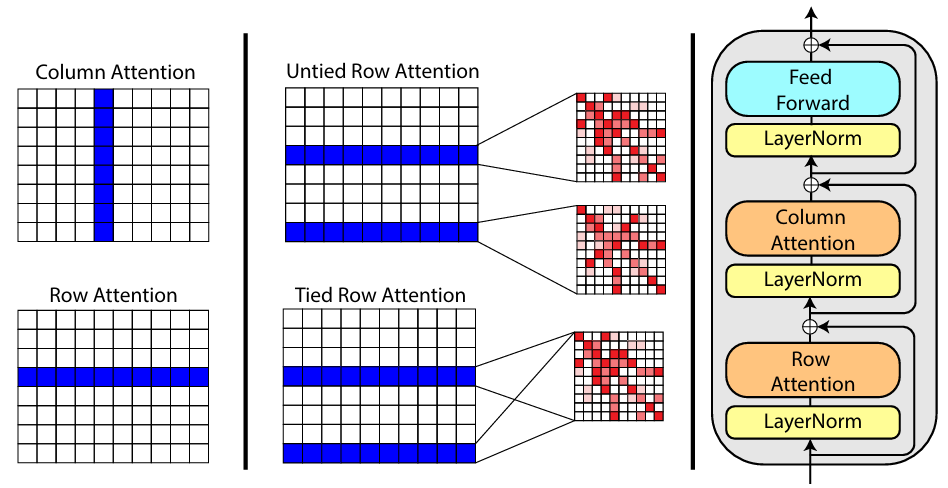
Il existe de nombreuses séquences menant au succès et de nombreuses séquences menant à l’échec.
L’AMS est une mine d’informations sur l’évolution, mais les modèles existants semblent surestimer leur capacité à la découvrir. Avec le développement de la technologie, la profondeur MSA des entrées de modèles génératifs profonds a continué d'augmenter, mais l'effet a rencontré un goulot d'étranglement, ce qui a remis en question la rentabilité de l'ajout d'informations MSA. La cause fondamentale est qu’il existe une sérieuse incertitude quant à la quantité et à la qualité des MSA :
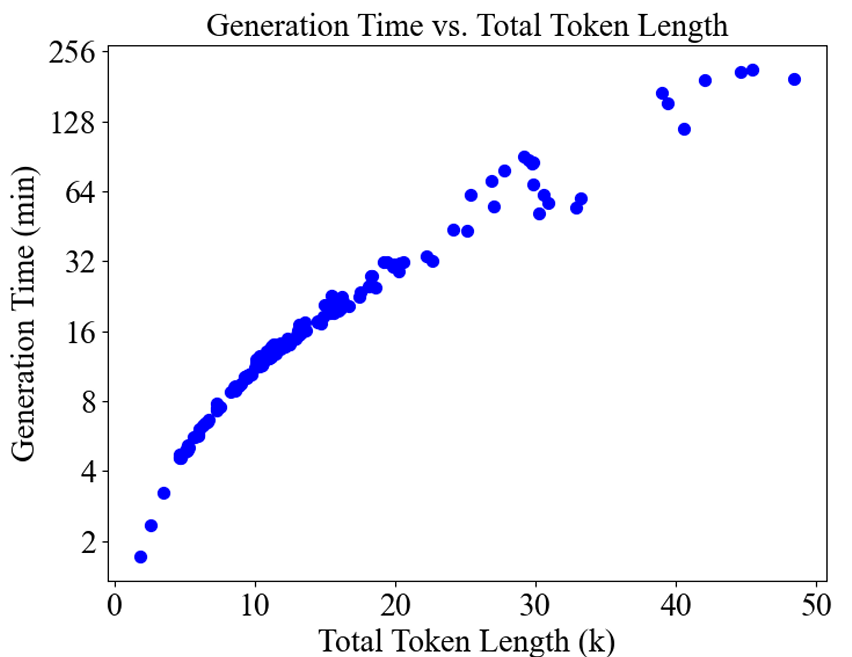
Les chercheurs appellent séquences homologues les séquences qui présentent un certain degré de similarité dans l’alignement de séquences multiples. En termes de quantité, pour certaines protéines « orphelines », il peut n'y avoir que 10 séquences homologues, tandis que pour certaines protéines, plus de 10 000 séquences homologues peuvent être recherchées, ce qui entraîne une grande confusion pour les grands modèles, entraînant un gaspillage de ressources et un impact sur l'efficacité.
En fait, les merveilles de la nature dépassent l’imagination humaine. Au cours de milliards d’années d’évolution, les structures convergentes reflètent les effets de la sélection naturelle, tandis que les mutations offrent de nouvelles possibilités d’évolution. Pour ces espèces particulières vivant dans des environnements particuliers, elles conservent souvent les informations d'apparence originales au début de l'arbre évolutif, ce qui constitue précisément la base de la déduction de la théorie de la coévolution. Si des séquences homologues sont utilisées comme entrée du modèle, ces informations seront forcément submergées par une grande quantité d'autres informations non pertinentes, et seules des représentations à haute probabilité pourront être modélisées. Pour remédier à cela,ProfileBFN modélise chaque groupe de séquences homologues comme une représentation unifiée indépendante du nombre.
Une bonne séquence homologue doit contenir autant d’informations homologues que possible. Les expériences montrent que dans la plupart des cas, l’utilisation de quelques séquences homologues avec la plus grande entropie d’information peut produire le même effet que l’utilisation de centaines de séquences homologues. Certaines séquences homologues ne diffèrent que de quelques acides aminés, ce qui fournit au modèle de nombreuses informations redondantes et trompeuses.
Profil : La pierre angulaire du modèle de piédestal protéique de nouvelle génération
La science est basée sur la découverte.L'innovation de ProfileBFN réside dans la découverte de la grande quantité de redondance d'informations existant dans le MSA d'origine. Si 100 séquences homologues sont triées selon la méthode d’entropie de l’information, le modèle peut obtenir le même effet en utilisant uniquement les 20 premières pour la formation. Pour ce faire, un pont entre une séquence unique et des séquences multiples doit être établi, c'est pourquoi le profil apparaît :
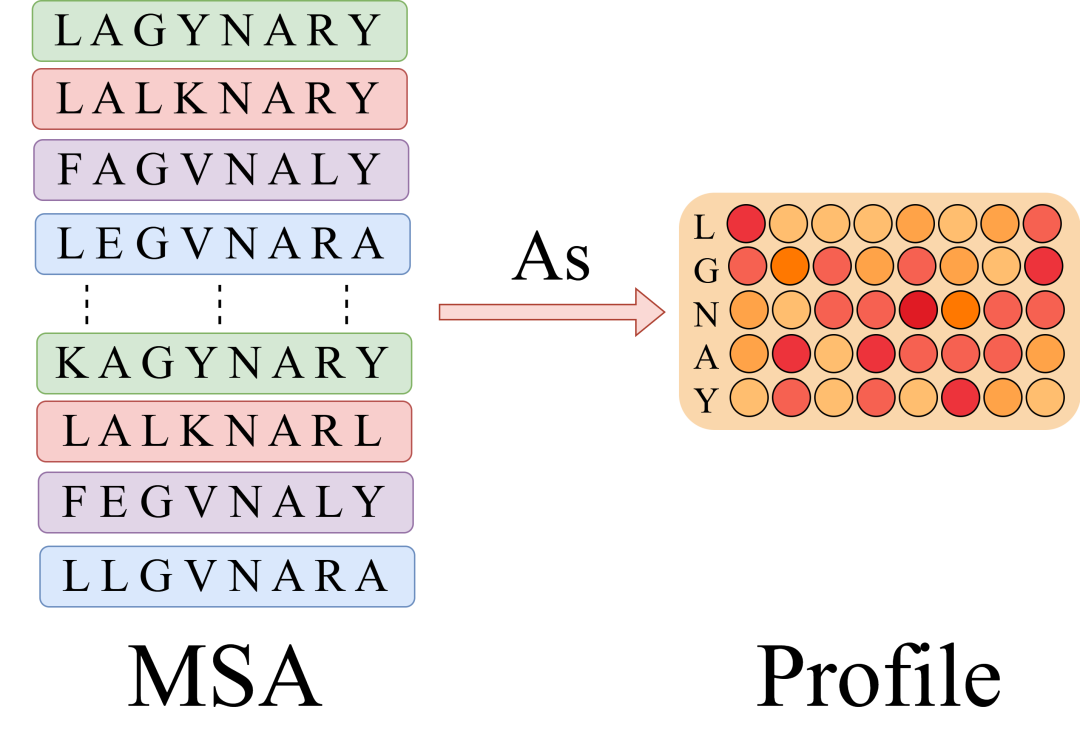
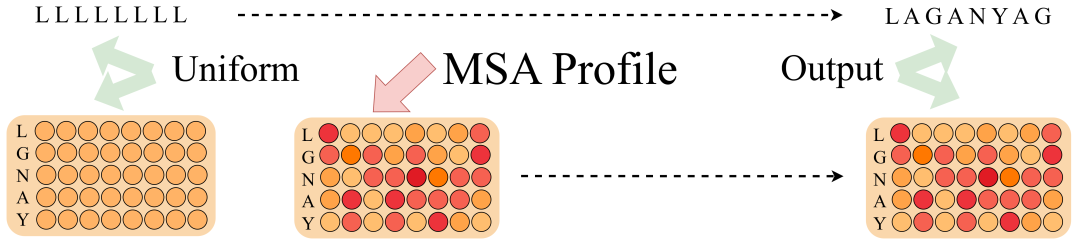
Pour comprendre intuitivement, le profil est une statistique colonne par colonne du nombre d'occurrences d'acides aminés dans un alignement de séquences multiples. De plus, s'il existe des séquences homologues de 1w, chacune d'une longueur de 100, Profile les compressera directement de [10000,100] à une liste de [20,100] (20 acides aminés communs), ce qui simplifie grandement la complexité du calcul. En particulier, une séquence unique peut également être considérée comme un profil spécial, sauf qu'il n'y a qu'un seul 1 dans chaque colonne.
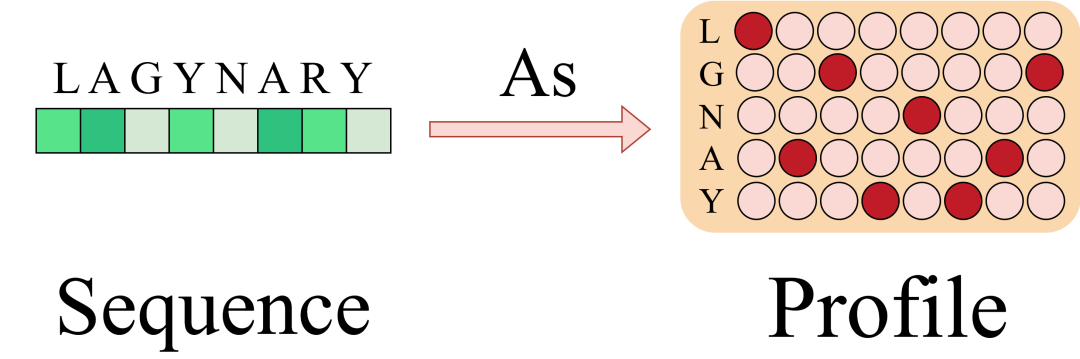
ProfileBFN a constaté que la compression de MSA vers Profile non seulement n'entraînait pas la perte d'informations grave initialement prévue, mais améliorait également considérablement les performances du modèle.Cela peut être compris comme : dans la grande vague de construction de Profil,Chaque séquence homologue vote sur le type d'acide aminé qui apparaît à cette position, masquant les contradictions mineures et mettant en évidence la tendance générale.
Les performances étonnamment fortes de ProfileBFN
Par rapport à la méthode traditionnelle basée sur l'alignement de séquences multiples,ProfileBFN s'appuie sur 10 fois moins de données et apprend 1,5 fois plus d'informations contextuelles sur les séquences de protéines.L'effet est immédiat !
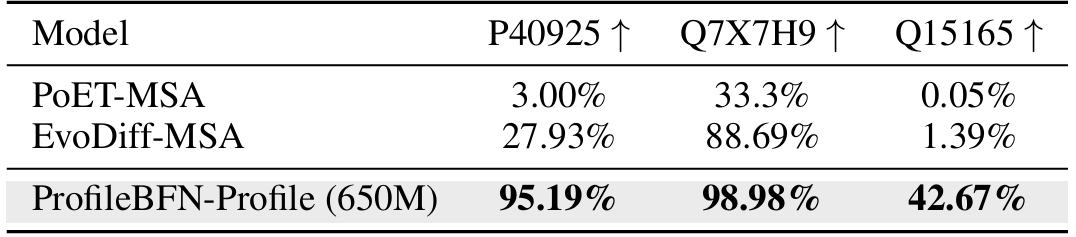
Après exploration, il a été confirmé que ProfileBFN a un effet facilitateur sur une variété de tâches en aval :
* Classification des enzymes :Améliorer la fidélité fonctionnelle et réduire les coûts de dépistage
* Apprentissage de la représentation des protéines :Aide à l'extraction de fonctionnalités multitâches
* Prédiction de la structure des protéines :Améliorer les informations d'homologie et améliorer la précision de la modélisation
* Production d'anticorps :Excellent effet de migration, prédiction précise des zones fonctionnelles
Les enzymes sont une classe spéciale de protéines dotées d'une activité catalytique, et leur spécificité fonctionnelle est généralement décrite par des numéros EC (Enzyme Commission Numbers). L'étude a révélé que les nouveaux candidats enzymatiques générés par ProfileBFN correspondaient fortement à l'enzyme de type sauvage en termes de nombre d'EC, ce qui signifie que les protéines générées maintenaient un degré élevé de cohérence fonctionnelle. Cette fonctionnalité réduit considérablement la difficulté du criblage expérimental et améliore le taux de réussite de la conception de nouvelles enzymes.
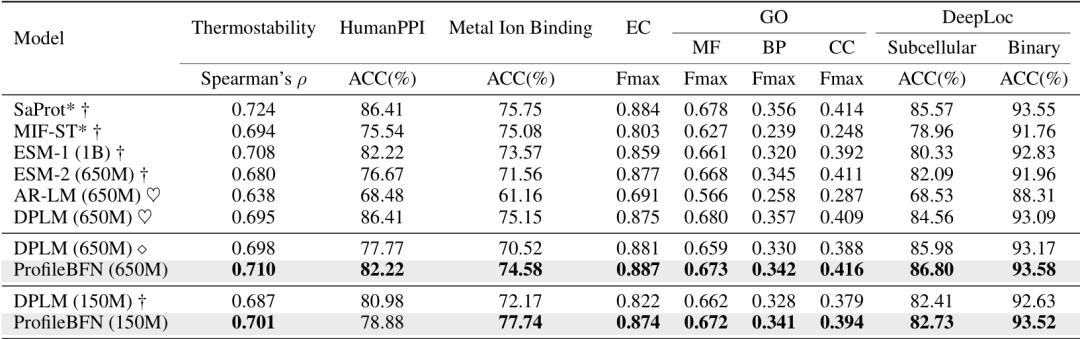
Alors que ProfileBFN génère des protéines, il crée également des représentations protéiques précises au sein du modèle. Les chercheurs ont extrait ces représentations,Il a été affiné sur plusieurs ensembles de données tels que la stabilité thermique des protéines, l’interaction des protéines et la localisation subcellulaire des protéines. Les résultats ont montré que la représentation fournie par ProfileBFN peut améliorer efficacement les performances du modèle dans les tâches en aval telles que la classification. Cela suggère qu’il ne s’agit pas seulement d’un modèle génératif, mais également d’un puissant outil d’apprentissage des fonctionnalités.
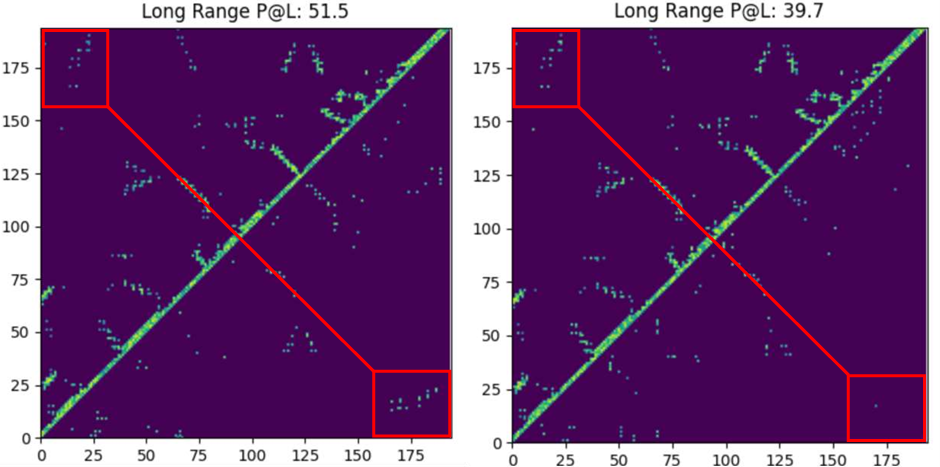
La prédiction de la structure des protéines est une question importante en biologie structurale.En particulier dans le cas des protéines orphelines (c’est-à-dire des protéines avec très peu de protéines homologues), la précision des méthodes traditionnelles est très limitée. Des études ont montré que ProfileBFN peut être utilisé comme un amplificateur d’informations d’homologie.Avec seulement une petite quantité de données MSA, davantage de protéines homologues de haute qualité sont générées, améliorant ainsi la précision de prédiction des modèles de la série AlphaFold. Cette capacité offre à ProfileBFN de larges perspectives d’application dans le domaine de la biologie structurale.
Les anticorps sont des protéines fonctionnelles qui peuvent se lier spécifiquement aux antigènes et sont d’une grande importance dans la recherche immunitaire et pathologique. Pour explorer le potentiel de ProfileBFN dans la génération d'anticorps,Les chercheurs ont affiné le modèle en se basant sur la base de données de séquences d’anticorps OAS (Observed Antibody Space).Les résultats ont montré que ProfileBFN était performant dans la génération de séquences d’anticorps diverses et de haute qualité.
L'effet extraordinaire de ProfileBFN vient du fait que cette nouvelle recherche fournit un paradigme pour générer des séquences biologiques à l'ère post-MSA :
* MSA ne participe pas directement au processus de formation en tant que contribution et n'introduit pas de frais de formation supplémentaires
* Dans la phase d'inférence, la séquence unique et l'AMS sont modélisées uniformément
* Les séquences homologues sont à la fois une entrée et une sortie du modèle
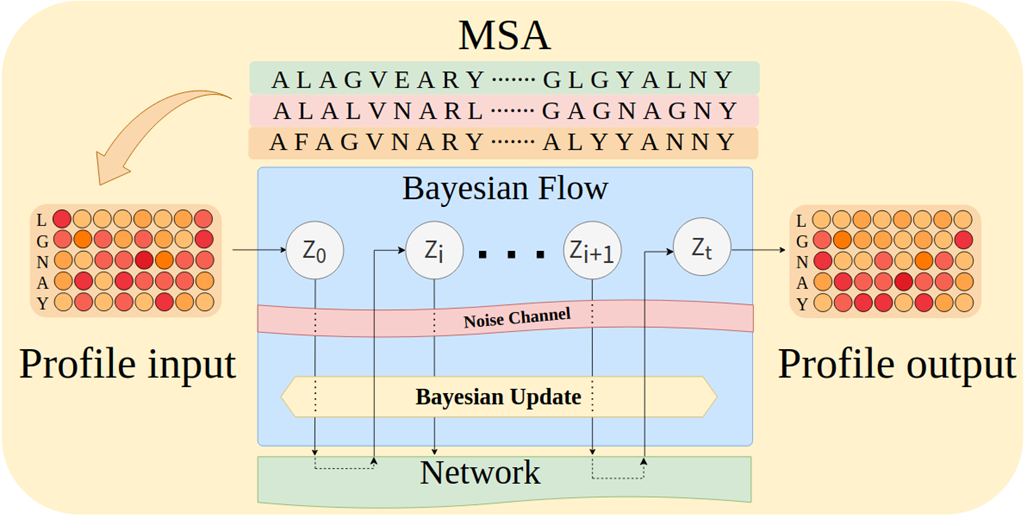
BFN utilise parfaitement les informations préalables
Étant donné que les informations de profil sont très importantes, encore plus importantes que la séquence homologue d'origine, comment devrions-nous utiliser les informations de profil ? Le réseau de flux bayésien BFN correspond parfaitement à Profile ! Cela se reflète dans deux points :
* BFN modélise le processus de distribution à distribution, l'entrée est une représentation de profil et la sortie est toujours une représentation de profil
* Au lieu de raisonner à partir de zéro, BFN peut introduire des informations de profil comme a priori pour le raisonnement conditionnel
Les modèles traditionnels tels que le modèle autorégressif et le modèle de diffusion nécessitent des données (jetons) en entrée, et le traitement des informations de profil augmentera la complexité de l'algorithme.
Avec BFN comme squelette de modèle, ProfileBFN peut en outre réaliser :
* Simplification des tâches. La génération conditionnelle d'informations homologues devient une imitation d'informations de profil.
* Efficacité améliorée. La plage d'échantillonnage est réduite et l'efficacité est améliorée
ProfileBFN devrait être le sauveur des tests humides
Dans des tâches telles que la biologie synthétique, les cycles longs, les indicateurs d’évaluation uniques et le manque de crédibilité sont des problèmes courants rencontrés par les chercheurs. En tant que modèle de base protéique, ProfileBFN peut intégrer davantage d'informations homologues avec des ressources limitées, exploiter pleinement des informations préalables spécifiques et avoir un bon effet de migration sur plusieurs indicateurs, ce qui en fait sans aucun doute le meilleur choix pour la synthèse de protéines candidates et l'évolution dirigée.
À propos du groupe de recherche
Le domaine de recherche du groupe de recherche sur l'intelligence symbolique générative (GenSI) de l'Institut de l'industrie intelligente de l'Université Tsinghua couvre les deux directions du LLM et de l'IA pour la science. On s’attend à ce que les deux directions se favorisent mutuellement et atteignent ainsi la mission ultime de l’AGI pour la science (AI Scientist).
Les axes de recherche spécifiques comprennent la nouvelle génération de technologie de pré-formation à grande échelle, l'apprentissage par renforcement à grande échelle (Large Scale RL), les modèles génératifs profonds (Deep Generative Models) et leurs applications dans les données scientifiques, en se concentrant sur l'intégration et l'innovation des algorithmes d'intelligence artificielle de base et des problèmes scientifiques. Actuellement, l'équipe se concentre sur les théories de pointe des modèles génératifs profonds et sur l'exploration des méthodes de modèles génératifs structurés évolutifs, et s'engage à résoudre des problèmes scientifiques réalistes et stimulants dans les domaines du LLM et de l'AI4Sci, tels que l'amélioration de la capacité de raisonnement du LLM et le dépassement des tâches de génération de structure de niveau AF3.
L'équipe peut être contactée via les canaux suivants⬇️
* page d'accueil:https://go.hyper.ai/7ye91
* Courriel : [email protected]
* Xiaohongshu/Zhihu : GenSI
* Twitter : @GenSI_official
* WeChat : 15805171115