Command Palette
Search for a command to run...
Prédiction Des Paramètres Cinétiques Enzymatiques, Identification Des Goulots d'étranglement... Luo Xiaozhou De l'Institut De Technologie Avancée De Shenzhen, Académie Chinoise Des Sciences, a Partagé l'application Innovante De l'IA Dans Le Domaine Des Enzymes

Les protéines, en tant que pierre angulaire de la vie, jouent un rôle clé dans les activités de la vie. L’étude de sa structure et de sa fonction est d’une grande importance pour le développement de médicaments innovants, la biologie synthétique, la production d’enzymes et d’autres domaines. Cependant, la conception traditionnelle des protéines est confrontée à de nombreux défis. La structure des protéines est complexe et l’espace de séquence est immense. La méthode de conception qui s’appuie sur l’expérience d’experts et sur un criblage à haut débit est non seulement longue et exigeante en main-d’œuvre, mais elle nécessite également des efforts pour garantir un taux de réussite difficile.
Aujourd’hui, l’IA pour la science est devenue une nouvelle frontière dans le développement de l’intelligence artificielle mondiale, qui change profondément le paradigme de la recherche scientifique et entraîne d’énormes changements dans le domaine de la conception des protéines. Surtout après l’émergence de résultats innovants tels qu’AlphaFold, les recherches connexes sont progressivement devenues publiques et ont reçu davantage d’attention. Dans le même temps, elle a également encouragé davantage d'équipes exceptionnelles au pays et à l'étranger à se consacrer à ce domaine et à relever les difficultés sous différents aspects tels que la technologie et l'application.
Le professeur Luo Xiaozhou, chercheur à l’Institut de technologie avancée de Shenzhen de l’Académie chinoise des sciences, est l’un d’entre eux. Il s’est auparavant concentré sur la biologie synthétique. Après son retour en Chine en 2019, il a commencé à se consacrer à la recherche sur les protéines de l'IA. Lors du sommet sur la conception de protéines IA « Future is Here » récemment organisé par l'Université Jiao Tong de Shanghai en Chine, le professeur Luo Xiaozhou a partagé son point de vue sur le thème de « l'ingénierie enzymatique basée sur l'intelligence artificielle ». Explorer les applications potentielles de l'apprentissage multimodal et de l'IA générative dans la conception d'enzymes,Les applications et pratiques innovantes de l'IA dans le domaine de l'ingénierie enzymatique sont expliquées sous de multiples perspectives telles que le cadre UniKP et la machine ProEnsemble.

HyperAI a organisé et résumé le partage approfondi sans violer l'intention initiale. Voici une transcription des points saillants du discours.
Construction de plateformes automatisées, l'IA résout les problèmes de protéines
Les produits naturels sont un véritable trésor de substances médicinales, avec les caractéristiques de sources étendues, de structures riches et d’activités diverses. Cependant, la méthode traditionnelle d’extraction de produits naturels à partir de ressources naturelles est inefficace et la synthèse chimique pure a non seulement de faibles rendements mais nécessite également l’utilisation d’une grande quantité de réactifs toxiques et dangereux. Par exemple, l’artémisinine était à l’origine extraite de l’Artemisia annua, mais elle a rencontré de nombreux problèmes lors de la synthèse chimique. Plus tard, l’expression de l’artémisinine a été obtenue dans Saccharomyces cerevisiae en régulant plusieurs gènes. Cette percée nous a permis de voir le potentiel de la biosynthèse, j’ai donc commencé à me concentrer sur la recherche dans le domaine biologique. De plus, dans le domaine de la modification enzymatique, le manque de données limitera sérieusement les progrès de la recherche. Ce problème nous fait prendre conscience de l’importance des données, c’est pourquoi je m’engage à créer des plateformes d’automatisation et de données pour jeter les bases des recherches ultérieures en IA.
En tant que molécules de base de la vie, les acides nucléiques, les lipides à petites molécules, les glucides, les métabolites, les ions, l’eau et d’autres substances sont tous produits à partir de protéines. Sur la base de cette caractéristique, après mon retour en Chine en 2019, j'ai concentré mes recherches sur le domaine des protéines et soulevé trois questions scientifiques : Premièrement, est-il possible de prédire l'activité et la fonction d'une protéine directement à partir de sa séquence ? La deuxième question est de savoir s’il est possible de générer ou de faire évoluer les protéines dont les gens ont besoin à la demande ? La troisième question est de savoir s’il est possible d’optimiser les enzymes ou les souches sur la base d’une stratégie universelle et standardisée ?
Le cadre UniKP prédit mieux les propriétés des enzymes
Le manuel stipule : La séquence primaire d’une protéine détermine sa structure tertiaire et sa fonction, et la séquence primaire doit contenir des informations fonctionnelles. Par conséquent, la manière d’extraire la séquence est extrêmement critique. Inspirée par AlphaFold, notre équipe a commencé à explorer des méthodes pour prédire la fonction des protéines à partir de la séquence. Dans notre étude, nous avons introduit l’architecture Transformer pour intégrer les méthodes de représentation traditionnelles avec les fonctionnalités d’apprentissage automatique afin de créer un modèle intégré.Le cadre de prédiction des fonctions des peptides et des protéines basé sur des caractéristiques de fusion et des modèles intégrés a atteint des performances SOTA sur 8 tâches de prédiction associées, prédisant avec précision les fonctions des peptides et des protéines.Il accélère le processus de criblage de substances actives anti-infectieuses telles que les peptides antimicrobiens et réduit les coûts expérimentaux.
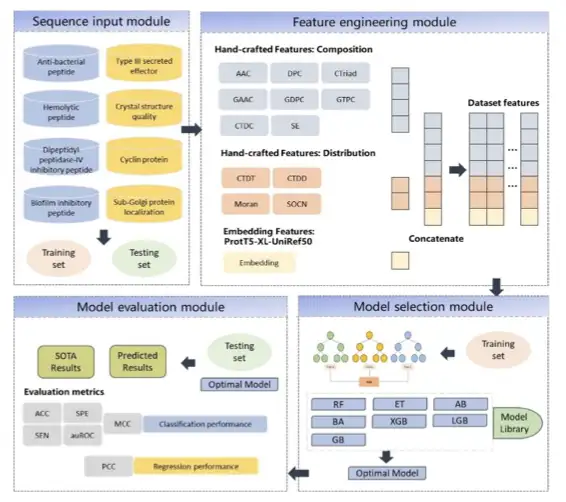
Par la suite, l'équipe a utilisé le cadre UniKP pour tenter de prédire les propriétés des enzymes en se basant sur l'outil de prédiction des paramètres enzymatiques d'intégration de Transformer. Utilisez ProtT5 et le modèle traditionnel SMILE Transformer pour vectoriser la séquence et la combiner avec un modèle d'apprentissage automatique simple pour obtenir des résultats SOTA.
L’équipe de recherche a sélectionné quatre ensembles de données représentatifs pour vérifier les performances et la valeur d’UniKP.
Le premier est l’ensemble de données DLkcat,Les chercheurs ont examiné 16 838 échantillons, dont 7 822 séquences protéiques uniques et 2 672 substrats uniques provenant de 851 organismes. L'ensemble de données est divisé en un ensemble d'entraînement et un ensemble de test dans un rapport de 9:1.
Viennent ensuite les ensembles de données de pH et de température,L'ensemble de données de pH contient 636 échantillons, constitués de 261 séquences enzymatiques uniques et de 331 substrats uniques ; L'ensemble de données de température contient 572 échantillons, constitués de 243 séquences enzymatiques uniques et de 302 substrats uniques. L'ensemble de données est divisé en un ensemble d'entraînement et un ensemble de test dans un rapport de 8:2.
Le troisième est l’ensemble de données de la constante de Michaelis (Km),Il se compose de 11 722 échantillons, dont des séquences enzymatiques, des empreintes moléculaires de substrat et des valeurs Km correspondantes. L'ensemble de données est divisé en un ensemble d'entraînement et un ensemble de test dans un rapport de 8:2.
Le quatrième est l’ensemble de données kcat/Km,Contient 910 échantillons constitués de séquences enzymatiques, de structures de substrat et de leurs valeurs kcat/Km correspondantes.
Il a été vérifié qu'UniKP est significativement meilleur que les modèles existants dans la prédiction kcat et atteint pour la première fois la prédiction kcat/Km.Prenons l’exemple de kcat, sur le plus grand ensemble de données accessible au public, le coefficient de détermination est supérieur de 20 points de pourcentage au résultat SOTA actuel. En même temps, il fonctionne également de manière significativement meilleure dans plusieurs tâches telles que différentes divisions d’ensembles de données, différentes divisions d’intervalles et différentes divisions de catégories d’enzymes.
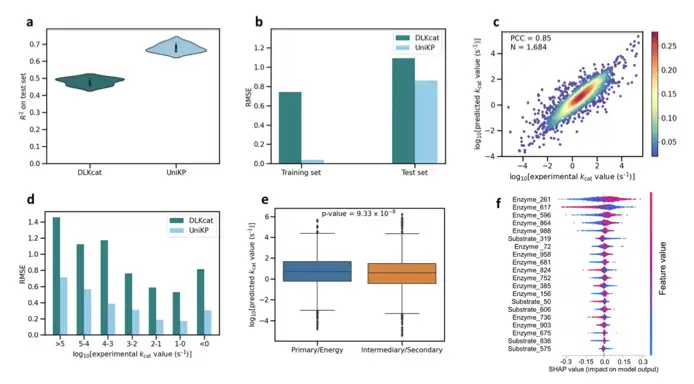
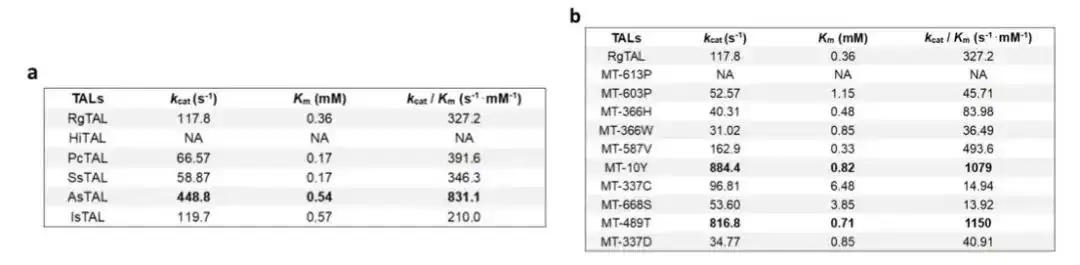
En utilisant cette architecture, nous avons trouvé l'enzyme TAL de type sauvage avec l'activité enzymatique la plus élevée à ce jour parmi 1 000 séquences Blast, et avons obtenu des mutants avec une activité enzymatique plus élevée en prédisant des mutations à site unique, accélérant considérablement le processus d'ingénierie enzymatique.
De plus, en ciblant la stabilité thermique des protéines, nous avons proposé un modèle de prédiction de protéines thermophiles basé sur la séquence Thermal Finer, qui a obtenu des performances SOTA sur trois ensembles de données de classification et a pour la première fois réussi à prédire la température catalytique optimale correspondante (régression) basée sur la séquence protéique. En d’autres termes, nous avons réussi pour la première fois à prédire directement la température optimale à partir de la séquence protéique, ce qui constitue un support solide pour l’exploration et l’évolution des enzymes.
Réglage fin de ProGPT-2 pour générer ou faire évoluer des protéines à la demande
Actuellement, il existe deux principaux types de modèles de production de protéines, notamment de production d’enzymes :
* Réseaux neuronaux antagonistes génératifs (GAN) : ProteinGAN
* Modèles de langage génératifs pré-entraînés (LLM) : ProtGPT2, ProGen
mais,Ces outils de génération de protéines ont tous pour problème de générer des séquences similaires et sont incapables de répondre aux besoins de génération d’enzymes avec de nouvelles fonctions et de nouvelles activités.Il y a aussi quelques aspects déraisonnables dans l'analyse théorique : premièrement, les valeurs des pixels de l'image sont continues, ce qui est plus adapté à l'optimisation du gradient ; deuxièmement, le texte (séquence d'acides aminés) est discontinu, et l'optimisation du gradient n'a aucun sens pour la mise à jour des intégrations, et elle est très inefficace.
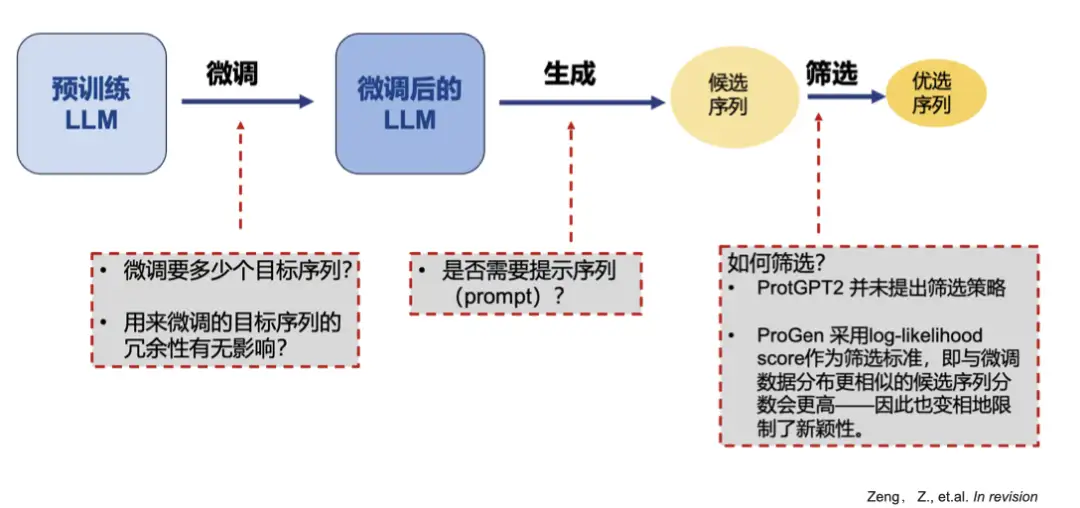
Pour de tels problèmes, nous analysons en profondeur les lacunes des modèles existants et proposons un nouveau cadre d’optimisation.
Notre équipe a peaufiné ProGPT-2 et utilisé un réseau neuronal CNN comme discriminateur pour filtrer et hiérarchiser les séquences générées. Grâce à des expériences, il a été découvert queLe réglage fin de la séquence ne nécessite que 2000 mots, voire moins, et la séquence générée sans mots d'indice est plus proche de l'enzyme naturelle. Dans le même temps, la réduction des données redondantes peut améliorer la nouveauté des séquences générées.
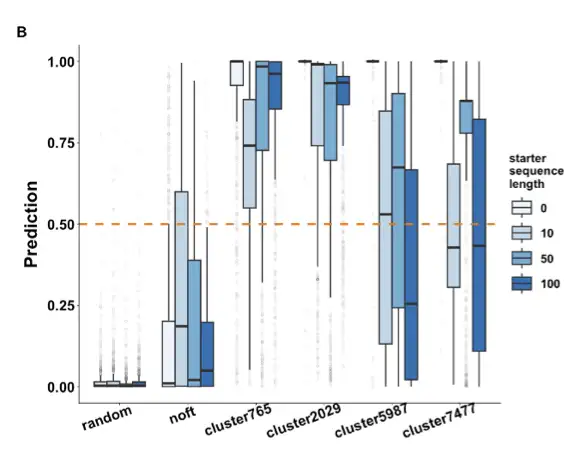
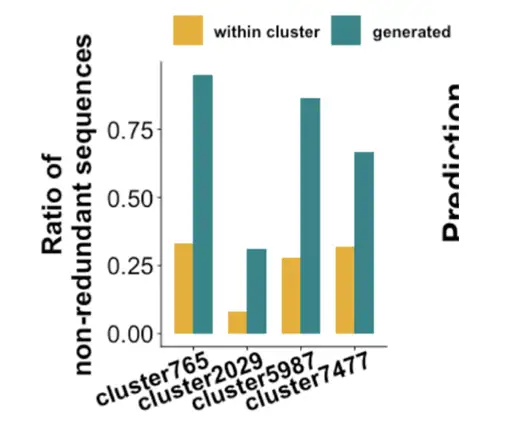
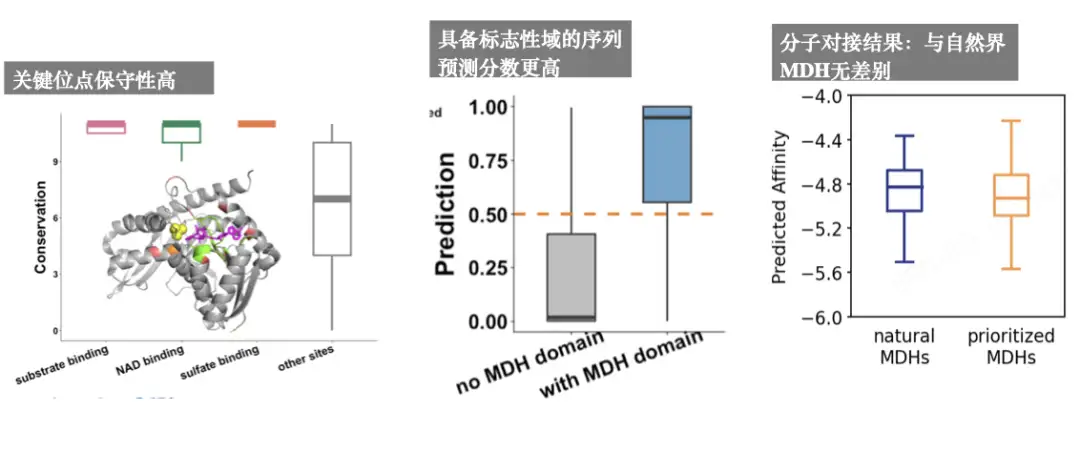
Nous voulons de nouvelles enzymes avec de nouvelles structures et fonctions, nous n’avons donc pas besoin de séquences redondantes. Grâce à des peptides antimicrobiens prédictifs, nous avons constaté que le modèle fonctionnait globalement bien, puis nous avons effectué une analyse MDH et avons constaté que :Les sites clés sont hautement conservés ; les scores de prédiction de ceux qui ont des domaines de signature sont plus élevés ; et les résultats de l'amarrage moléculaire sont fondamentalement les mêmes que ceux du MDH dans la nature.Comme le montre la figure suivante :
Nous avons ensuite vérifié si les enzymes différentes produites après le modèle étaient fonctionnelles. Sur la base des données originales de ProteinGAN, les enzymes présentant une similarité de 80% peuvent atteindre une similarité inférieure à 40% après le modèle MDHs prioritaire. Comparé aux 10 enzymes que nous avons sélectionnées au hasard dans la nature, il est fondamentalement le même en termes d'insoluble, sans expression et soluble, mais il a toujours une très bonne activité enzymatique. Autrement dit,Les enzymes générées par notre équipe à l’aide de ce modèle présentent une faible similitude avec les enzymes naturelles et la plupart d’entre elles ont une activité enzymatique.
ProEnsemble Identifiez les goulots d'étranglement métaboliques et optimisez la production d'enzymes
Dans le processus de biosynthèse, une série de goulots d'étranglement métaboliques tels que la faible efficacité catalytique de plusieurs enzymes dans la voie métabolique et les effets épistatiques entre les enzymes rendent le processus d'optimisation complexe et incertain. La surexpression des enzymes de la voie affecte souvent la croissance cellulaire et l’expression du produit, et certaines enzymes peuvent provoquer des effets négatifs. À cette fin, j’ai demandé s’il existe une stratégie universelle et standardisée pour optimiser les enzymes ou les souches ?
Vérifions d’abord si la surexpression est vraiment mauvaise ?L’équipe a réduit artificiellement les niveaux d’expression de certaines enzymes pour créer des goulots d’étranglement métaboliques artificiels, obtenant ainsi un espace évolutif contrôlable.
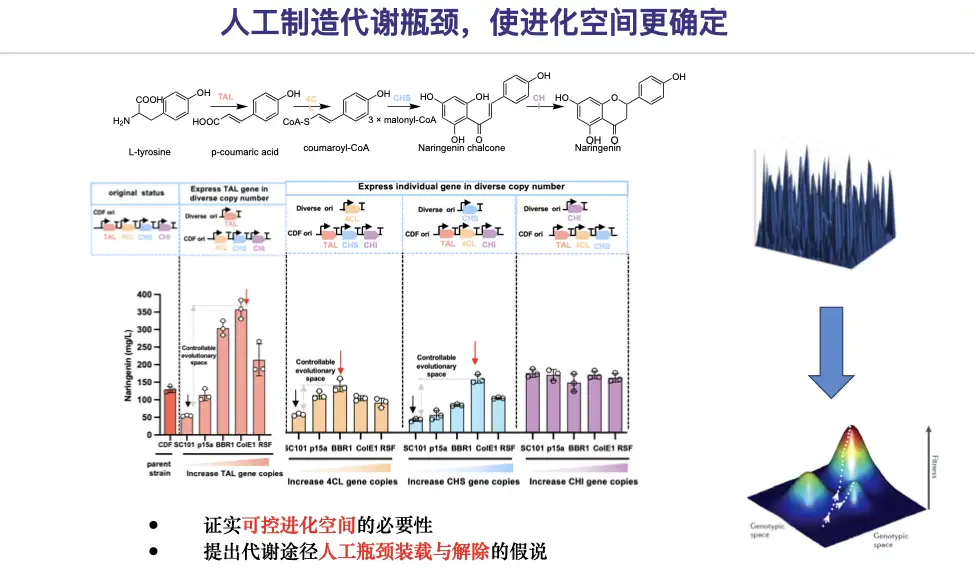
Par conséquent, une solution pour la conception des goulots d'étranglement des voies et une stratégie d'élimination est proposée, en prenant la naringénine comme exemple :
* Dans la première étape, nous utiliserons la technologie de la plate-forme automatisée à grande échelle pour permettre aux gènes liés à la synthèse de la naringénine d'être exprimés à un faible niveau (faible nombre de copies de fond), construisant ainsi un goulot d'étranglement métabolique artificiel pour la synthèse de la naringénine.
* Dans la deuxième étape, les mutants candidats 4CL-11C1 et CHS-9H9 ont été examinés pour leur production de naringénine comparable à celle des mutants originaux, éliminant ainsi le goulot d'étranglement de la voie de la naringénine.
* Dans la troisième étape, grâce à l’ingénierie des promoteurs médiée par l’IA, les mutants de gènes uniques sont replacés dans la voie d’origine et le flux métabolique est équilibré.
Les résultats de l’étude ont montré queLes stratégies de création et de suppression artificielles de goulots d’étranglement permettent une évolution efficace des voies métaboliques dans les limites de trajectoires claires.Cela confirme également que les effets épistatiques peuvent limiter les limites de l’évolution des voies.
Sur cette base, nous avons conçu un processus automatisé, comprenant des instructions, un clonage et des tests de dépistage bactérien.Les résultats ont montré qu’il n’y avait pas de différence significative entre cette méthode et l’opération manuelle en termes de croissance, de criblage et d’extraction du produit.Cependant, les méthodes d'évolution des voies métaboliques assistées par l'automatisationLe temps nécessaire à l’évolution parallèle de plusieurs enzymes est considérablement réduit et un cycle d’évolution parallèle peut être réalisé en deux semaines.
Sur la base d'une grande quantité de données accumulées, l'équipe a développé un modèle intégré d'apprentissage automatique ProEnsemble pour optimiser les incréments métaboliques. Les expériences ont montré que le modèle intégré basé sur l'apprentissage automatique équilibrait les voies métaboliques et augmentait la production de naringénine de 5,16 fois par rapport au modèle non optimisé, atteignant 1,21 g/L dans une plaque à 96 puits et 3,65 g/L dans un fermenteur, atteignant le niveau le plus élevé signalé. En surexprimant simplement les gènes synthétiques clés, la production de divers châssis composés modifiés était supérieure aux niveaux rapportés dans la littérature (avec l'aide de stratégies d'ingénierie métabolique).
La stratégie d'apprentissage ProEnsemble construit un système en boucle fermée d'identification et d'optimisation des goulots d'étranglement métaboliques, développant avec succès un châssis Escherichia coli à naringénine à haut rendement, qui est plusieurs fois supérieur au niveau actuel de l'industrie et fournit une solution universelle pour l'équilibre des réseaux métaboliques complexes.
Construire une plateforme d'automatisation à grande échelle pour promouvoir la coopération industrie-université-recherche
Enfin, je voudrais vous présenter la mise en œuvre industrielle de ces réalisations. Nous avons construit une plate-forme entièrement automatisée à grande échelle - la principale installation scientifique et technologique pour la recherche en biologie synthétique à Shenzhen, en Chine, qui comprend une plate-forme automatisée à grande échelle couvrant plusieurs plates-formes telles que l'apprentissage de la conception, les tests synthétiques et les tests utilisateurs. La plateforme dispose de fonctions puissantes et peut effectuer un traitement de données standardisé et une conception expérimentale pour l'apprentissage automatique dans le cloud. Les robots peuvent aider à réaliser des opérations expérimentales. La vitesse de préparation et de détection du spectre est rapide et un échantillon peut être généré en seulement 10 secondes, permettant une détection à haut débit.
De plus, la plateforme fournit également une conception logicielle assistée automatisée, permettant aux utilisateurs de sélectionner directement les composants requis dans la bibliothèque de composants et de générer des instructions expérimentales. Nous avons maintenant coopéré avec de nombreuses industries et universités. Nous sommes la première plateforme du secteur à réaliser l'intégralité du processus d'automatisation de Streptomyces. Nous invitons tout le monde à coopérer avec nous.
À propos du professeur Luo Xiaozhou
Le professeur Luo Xiaozhou est chercheur, directeur de doctorat, directeur adjoint de l'Institut de biologie synthétique des Instituts de technologie avancée de Shenzhen, Académie chinoise des sciences. Il est un expert sélectionné du National Major Talent Project - Youth Project, CTO du National Biomanufacturing Industry Innovation Center et ingénieur en chef adjoint des procédés de la Shenzhen Major Science and Technology Facility for Synthetic Biology, Chine.
Il a obtenu son doctorat. en chimie du Scripps Research Institute en 2016 (superviseur : académicien Peter G. Schultz), puis a effectué des recherches postdoctorales à l'Université de Californie à Berkeley (co-superviseur : académicien Jay D Keasling). En 2019, il rejoint les Instituts de technologie avancée de Shenzhen, Académie chinoise des sciences. Il a été sélectionné pour le programme national des jeunes talents, les jeunes chercheurs exceptionnels de la province du Guangdong et les excellents jeunes chercheurs de la ville de Shenzhen.
Ses recherches portent sur les processus biochimiques dans les organismes vivants dans le domaine de la biologie synthétique, y compris l’évolution dirigée des enzymes, l’ingénierie des protéines, le criblage à haut débit et la biosynthèse totale de composés naturels et non naturels. En tant qu'auteur correspondant, il a publié 20 articles dans Nature Metabolism, Advanced Science, Nature Synthesis, Nature Communications, Angew. Chimie Int. Ed., etc., et un total de plus de 50 articles SCI, demandé plus de 30 brevets et autorisé 6.








